Clear Sky Science · de
NUP93 erleichtert den Kernimport von SOX2, um die Transkription von G3BP1 zu aktivieren, und beeinträchtigt die Gemcitabin-Wirksamkeit bei Bauchspeicheldrüsenkrebs
Warum diese Forschung wichtig ist
Bauchspeicheldrüsenkrebs zählt zu den tödlichsten Krebsarten, unter anderem weil er Chemotherapien wie Gemcitabin häufig widersteht. Diese Studie legt eine verborgene Kette molekularer Ereignisse in pankreatischen Tumorzellen offen, die ihnen hilft, genau die DNA-Schäden zu reparieren, die Gemcitabin eigentlich verursachen soll. Indem die Autoren diese Kette vom Zellrand bis zum genetischen Kern zurückverfolgen, zeigen sie eine neue Verwundbarkeit, die künftige Therapien ausnutzen könnten, um Chemotherapien wirkungsvoller zu machen.
Ein Torwächter am Zellkern
Jede Zelle bewahrt ihre DNA in einem geschützten Kompartiment, dem Zellkern, der mit winzigen Toren, den nukleären Poren, besetzt ist. Das Protein NUP93 ist ein wichtiges strukturelles Bauteil dieser Tore. Durch die Auswertung großer Krebsdatenbanken und die Untersuchung von Patientenproben fanden die Forschenden heraus, dass die NUP93-Spiegel im Pankreasduktaladenokarzinom deutlich höher liegen als im normalen Pankreasgewebe. Patienten mit höherer NUP93-Expression in ihren Tumoren zeigten tendenziell eine schlechtere Überlebensrate. Im Labor kultivierte pankreatische Tumorzellen erwiesen sich als besonders abhängig von NUP93: Bei Reduktion von NUP93 wuchsen diese Zellen langsamer und bildeten weniger Kolonien, was darauf hindeutet, dass NUP93 mit aggressivem Tumorverhalten verknüpft ist.
Wie Krebszellen der Chemotherapie entgehen
Gemcitabin wirkt, indem es sich während der DNA-Replikation in das Erbgut einbaut und so Fehler und Brüche auslöst, die die Teilung von Tumorzellen stoppen sollten. Krebszellen können jedoch Gegenmaßnahmen ergreifen, indem sie DNA-Reparatursysteme aktivieren. Beim Vergleich von Tumoren mit hohem und niedrigem NUP93-Spiegel zeigte sich, dass Gene, die an der DNA-Reparatur beteiligt sind, bei hoher NUP93-Expression stärker aktiv waren. In Zellversuchen machte die Herunterregulierung von NUP93 pankreatische Tumorzellen deutlich empfindlicher gegenüber Gemcitabin, während erhöhte NUP93-Spiegel sie schwerer töten ließen. Marker für DNA-Schäden stiegen bei Verlust von NUP93 und gingen bei Überexpression zurück; ein Comet-Assay, der gebrochene DNA visualisiert, bestätigte, dass Zellen ohne NUP93 nach Behandlung mehr Schäden ansammelten. In Mäusen wuchsen Tumoren ohne NUP93 langsamer und sprachen deutlich besser auf Gemcitabin an.
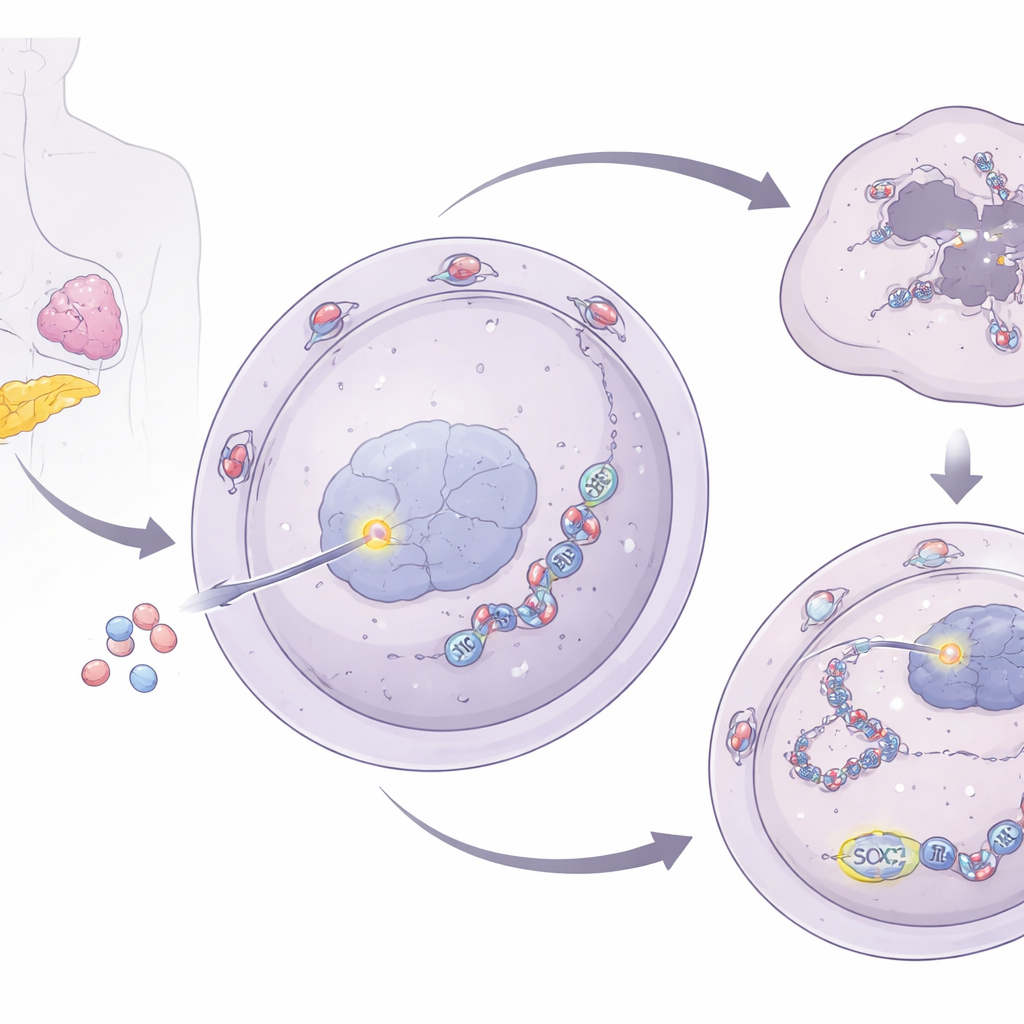
Ein Relay vom Kernporenkomplex zu Stressgranula
Um zu verstehen, wie NUP93 die DNA-Reparatur stärkt, untersuchten die Forschenden seine Verknüpfungen zu zellulären Stressgranula – temporären Clustern aus RNA und Proteinen, die Zellen helfen, widrige Bedingungen zu überstehen. Sie entdeckten, dass NUP93 die Spiegel von G3BP1, einem zentralen Stressgranula-Protein, erhöht. Bei gesteigerter NUP93-Expression stiegen auch G3BP1-Werte; bei Reduktion von NUP93 fielen G3BP1-Werte. Wichtig: Das Entfernen von G3BP1 hob NUP93s Fähigkeit auf, Zellwachstum und Gemcitabin-Resistenz zu fördern, und stellte die DNA-Schäden nach Behandlung wieder her. Das Team verfolgte die Kontrolle von G3BP1 weiter zurück zu SOX2, einem bekannten Transkriptionsfaktor, der Gene an- und abschaltet. NUP93 interagiert physisch mit SOX2 am Nukleopor und hilft, SOX2 in den Zellkern zu transportieren. Einmal im Kern bindet SOX2 direkt an die Regulatorregion des G3BP1-Gens und erhöht dessen Produktion.
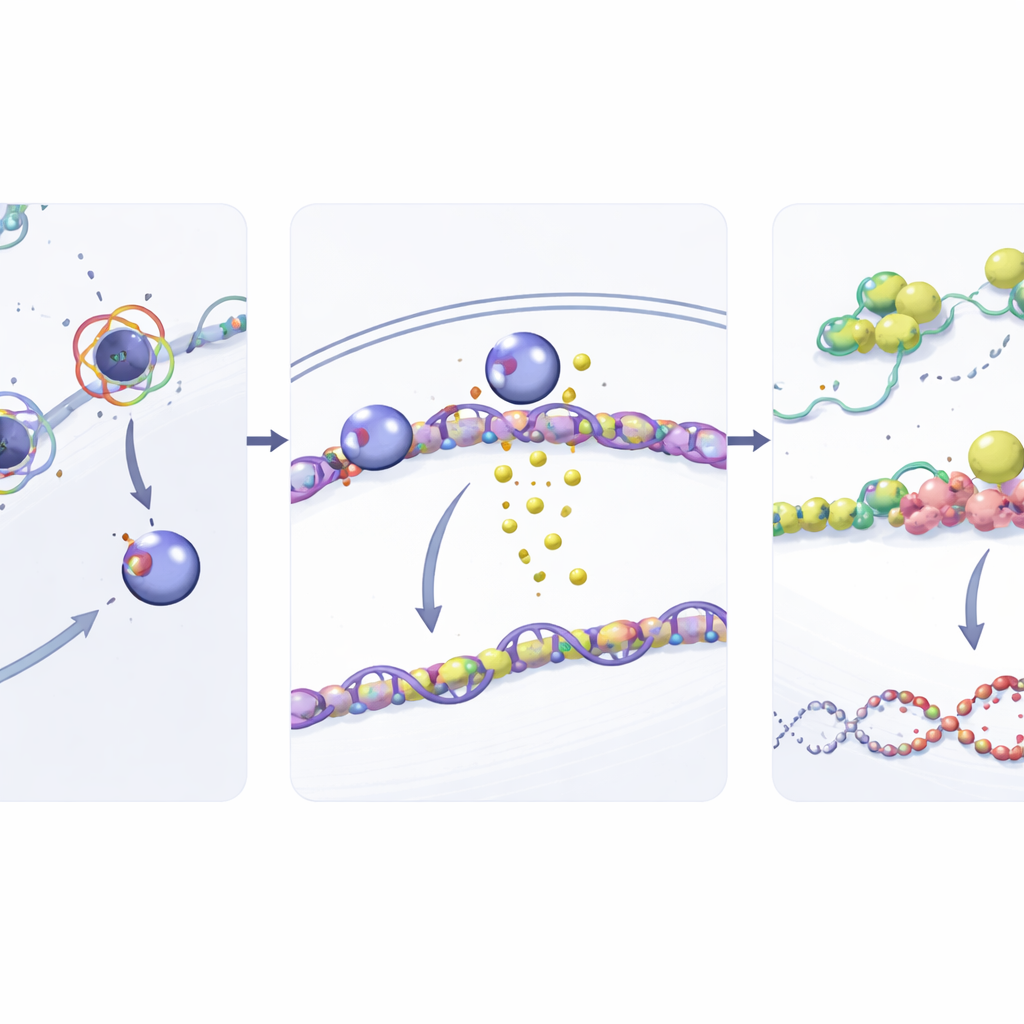
Von RNA-Schutz zur DNA-Reparatur
Die Geschichte endet nicht bei G3BP1. Die Forschenden zeigten, dass G3BP1 wiederum hilft, die Boten-RNA (mRNA) von RAD51 zu stabilisieren, einem zentralen Akteur im hochgenauen DNA-Reparaturweg der homologen Rekombination. G3BP1 bindet direkt an RAD51-mRNA und verlangsamt deren Abbau, was zu höheren RAD51-Proteinspiegeln führt. Mit mehr verfügbarer RAD51 sind Krebszellen besser gerüstet, gemcitabininduzierte DNA-Brüche zu reparieren. Bei Depletion von G3BP1 fielen die RAD51-Spiegel, RAD51-mRNA wurde schneller abgebaut und DNA-Schäden häuften sich an. In Mausmodellen verringerten Blockaden von SOX2 oder G3BP1 das Tumorwachstum und machten Gemcitabin deutlich wirksamer, was die Bedeutung dieses Relays in lebenden Systemen bestätigt.
Ein neues Ziel, um die Chemotherapie zu unterstützen
Setzt man die Teile zusammen, schlagen die Autoren einen klaren Weg vor: NUP93 am Nukleopor geleitet SOX2 in den Zellkern, SOX2 aktiviert G3BP1, und G3BP1 bewahrt RAD51-mRNA, wodurch schließlich die DNA-Reparatur verstärkt und pankreatische Tumorzellen gegenüber Gemcitabin resistenter werden. Diese NUP93–SOX2–G3BP1–RAD51-Achse wirkt wie ein internes Rettungsteam, das den Schaden repariert, den die Chemotherapie verursachen soll. Durch die Störung eines oder mehrerer Schritte dieser Kette könnten künftige Therapien die Abwehrkräfte der Krebszellen schwächen und vorhandene Medikamente wirkungsvoller machen — ein möglicher neuer Ansatz zur Verbesserung der Prognose bei dieser schwer behandelbaren Erkrankung.
Zitation: Sun, H., Yang, C., Du, J. et al. NUP93 facilitates the nuclear import of SOX2 to activate G3BP1 transcription and impairs gemcitabine response in pancreatic cancer. Cell Death Dis 17, 423 (2026). https://doi.org/10.1038/s41419-026-08586-4
Schlüsselwörter: Bauchspeicheldrüsenkrebs, Gemcitabin-Resistenz, DNA-Reparatur, nukleäres Porenkomplex, Stressgranula