Clear Sky Science · de
Osteoblastischer Sclerostin-Schleife3–LRP4‑Interaktion, die Sclerostin benötigt, um die Knochenbildung zu hemmen
Warum Knochenaufbau einen klügeren Schalter braucht
Brüchige Knochen und Frakturen sind ein wachsendes Problem, da die Menschen länger leben, und es gibt inzwischen potente neue Medikamente, die das Knochenwachstum anregen. Doch eines dieser Mittel – ein Antikörper, der ein knochenhemmendes Protein namens Sclerostin blockiert – wurde bei einigen Patienten mit schweren Herz-Kreislauf-Problemen in Verbindung gebracht. Diese Studie entschlüsselt die feinen Details, wie Sclerostin die Knochenbildung verlangsamt, und zeigt einen Weg auf, seine Bremse in Knochenzellen auszuschalten, während seine schützenden Funktionen im Herz-Kreislauf-System erhalten bleiben.
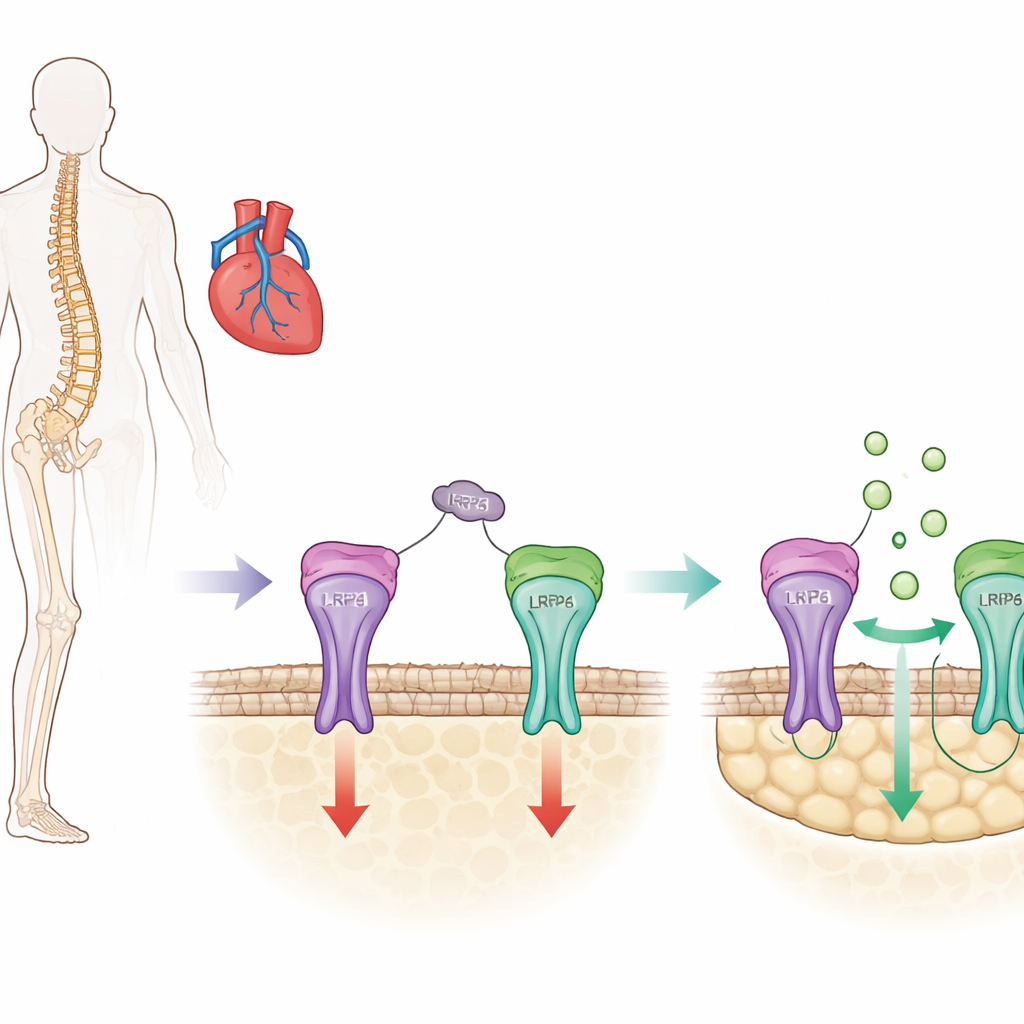
Ein wichtiger Knochen-Bremser mit versteckter Seite
Knochen werden ständig erneuert: spezialisierte Zellen bauen neues Gewebe auf, während andere altes Material entfernen. Sclerostin ist ein hauptsächlich von Knochenzellen gebildetes Protein, das als Bremse dieses Aufbauprozesses wirkt, indem es den wachstumsfördernden Wnt/β‑Catenin‑Signalweg in knochenbildenden Zellen (Osteoblasten) dämpft. Aktuelle Osteoporosemittel wie Romosozumab wirken, indem sie Sclerostin blockieren, diese Bremse lösen und so die Knochenmasse rasch erhöhen. Klinische Studien deuten jedoch darauf hin, dass eine breite Sclerostin‑Blockade auch schützende Funktionen in Gefäßen und Herz stören kann und so bei manchen Personen das Risiko für Herzinfarkt und Schlaganfall erhöht.
Heranzoomen an eine winzige Schleife
Sclerostin besitzt drei kleine, schleifenförmige Regionen in seiner Struktur; frühere Arbeiten dieser Gruppe zeigten, dass eine davon, genannt Schleife3, dabei hilft, die Knochenbildung zu unterdrücken, aber für die schützenden Effekte in Blutgefäßen nicht erforderlich ist. In der vorliegenden Studie fragten die Forscher genau, wie Schleife3 zur Knochenhemmung beiträgt. Sie entdeckten, dass Schleife3 an einen Rezeptor auf der Oberfläche der Knochenzelle bindet, LRP4 genannt. Diese Bindung wirkt wie ein Verankerungspunkt, der Sclerostin so positioniert, dass es einen benachbarten Rezeptor, LRP6, erreichen kann, der direkt den Wnt/β‑Catenin‑Weg steuert. Mithilfe biochemischer Bindungstests und Computermodellierung identifizierten sie bestimmte Aminosäuren im LRP4‑Protein, die essentielle Kontaktpunkte für Schleife3 darstellen.
Die Verankerung lösen, um das Knochenwachstum freizugeben
Ausgestattet mit diesen strukturellen Erkenntnissen entwickelte das Team zwei Werkzeuge, um die Schleife3–LRP4‑Verankerung zu stören: eine genetische Variante, die die Schlüsselaminosäuren in LRP4 subtil veränderte (Lrp4m genannt), und ein kurzes synthetisches Peptid (LRP4‑Pep), das den Schleife3‑bindenden Bereich von LRP4 nachahmt und mit Sclerostin konkurriert. In kultivierten Osteoblasten schwächten beide Strategien die Fähigkeit von Sclerostin, an LRP6 zu binden, verringerten seine Unterdrückung des Wnt/β‑Catenin‑Signals und erhielten die Reifung der Zellen sowie deren Mineralablagerung. Computersimulationen stützten dieses Bild: Normales LRP4 hält Sclerostin nahe an LRP6, während die mutierte Form diesen Kontakt nicht stabilisiert.
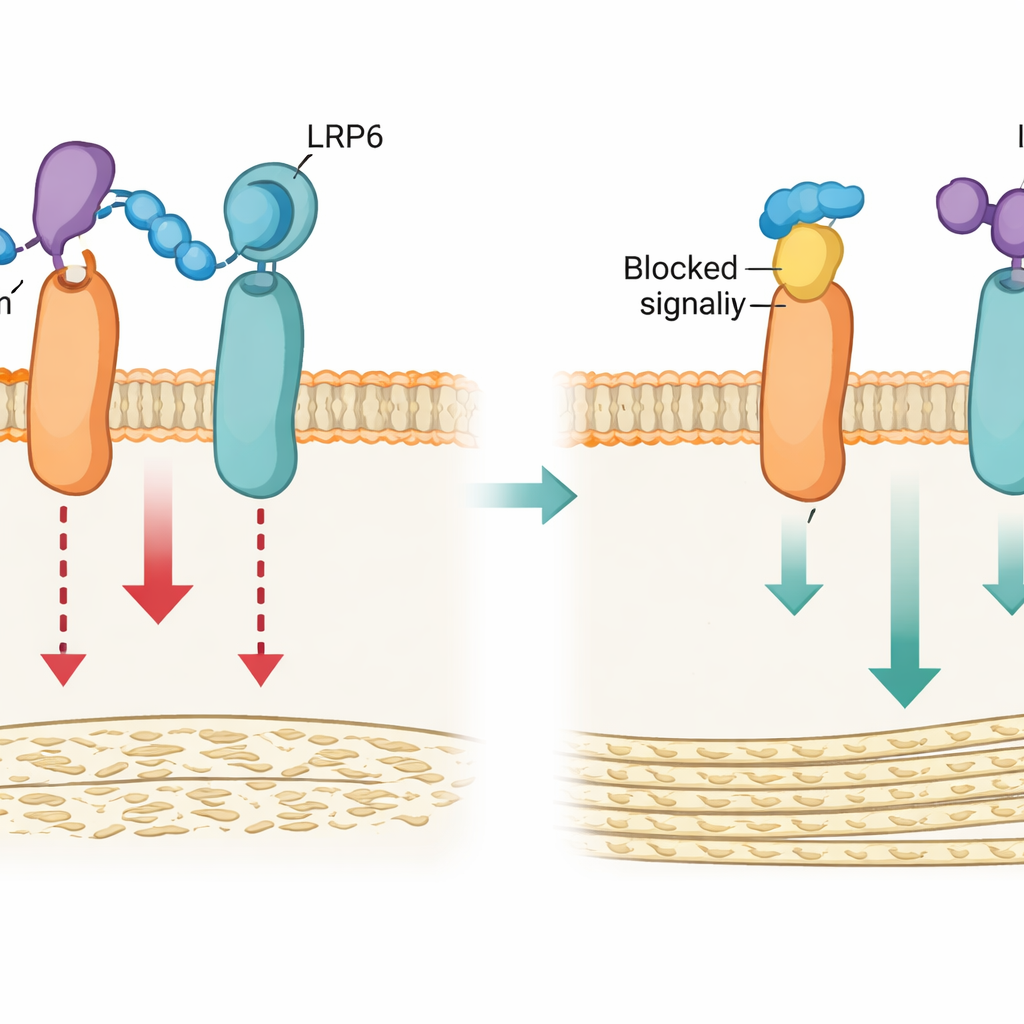
Stärkere Knochen bei Mäusen ohne zusätzliche Risiken
Anschließend prüften die Forscher Mausmodelle, um zu bestätigen, dass die Störung der Schleife3–LRP4‑Interaktion die Knochengesundheit in lebenden Tieren verbessert. Mäuse, die die Lrp4m‑Mutation im gesamten Körper trugen, entwickelten dichtere, stärkere Knochen mit verbesserter innerer Architektur und erhöhten Blutwerten von Knochenbildungsmarkern, ohne messbare Schädigung der Muskulatur. War die gleiche Mutation in Mäusen vorhanden, denen genetisch Sclerostin fehlte, zeigte sich kein zusätzlicher Effekt; die Wiedereinführung von Sclerostin aber machte deutlich, dass seine knochensuppressive Wirkung in Lrp4m‑Tieren deutlich geschwächt war. Ebenso führte wiederholte Behandlung mit dem LRP4‑Pep‑Peptid bei Mäusen, die zu viel Sclerostin produzierten, oder bei Mäusen mit etabliertem östrogenmangelbedingtem Knochenverlust zu dosisabhängigen Zunahmen der Knochenmasse, verbesserter Knochenqualität und erhöhter mechanischer Festigkeit, hatte jedoch keinen nachweisbaren Effekt, wenn Sclerostin nicht vorhanden war.
Auf dem Weg zu sichereren knochenanabolen Therapien
Zusammen zeigen diese Ergebnisse, dass die Interaktion zwischen Sclerostin‑Schleife3 und LRP4 in Osteoblasten die Verankerung darstellt, die Sclerostin benötigt, um sich an LRP6 anzuheften und die Knochenbildung abzuschalten. Durch das selektive Stören dieser Verankerung – entweder durch präzise Mutationen oder ein maßgeschneidertes Peptid – konnten die Forscher in Mäusen den Knochenaufbau und die Festigkeit steigern, ohne die Teile von Sclerostin direkt zu blockieren, die offenbar für den kardiovaskulären Schutz wichtig sind. Für Patientinnen und Patienten weist diese Arbeit auf eine neue Generation knochenstärkender Therapien hin, die nicht darauf zielen, Sclerostin vollständig stummzuschalten, sondern seine Effekte im Knochen von seinen nützlichen Funktionen im Herz‑Kreislauf‑System zu entkoppeln.
Zitation: Wang, L., Tao, X., Jiang, H. et al. Osteoblastic sclerostin loop3-LRP4 interaction required by sclerostin to inhibit bone formation. Bone Res 14, 45 (2026). https://doi.org/10.1038/s41413-026-00511-x
Schlüsselwörter: Osteoporose, Sclerostin, Knochenbildung, LRP4, Wnt-Signalgebung