Clear Sky Science · zh
探索用于粗粒化蛋白质折叠的量子退火
这对未来药物意味着什么
蛋白质折叠成的形状决定了它们在体内的功能,从运输氧气到识别病毒。预测这些形状对药物设计至关重要,但要准确预测所有蛋白质仍然极具挑战。本文探讨了一类新型计算硬件——量子退火器——是否有朝一日能够帮助解决那些对现有人工智能和经典超级计算机构成挑战的特别艰难的蛋白质折叠问题。
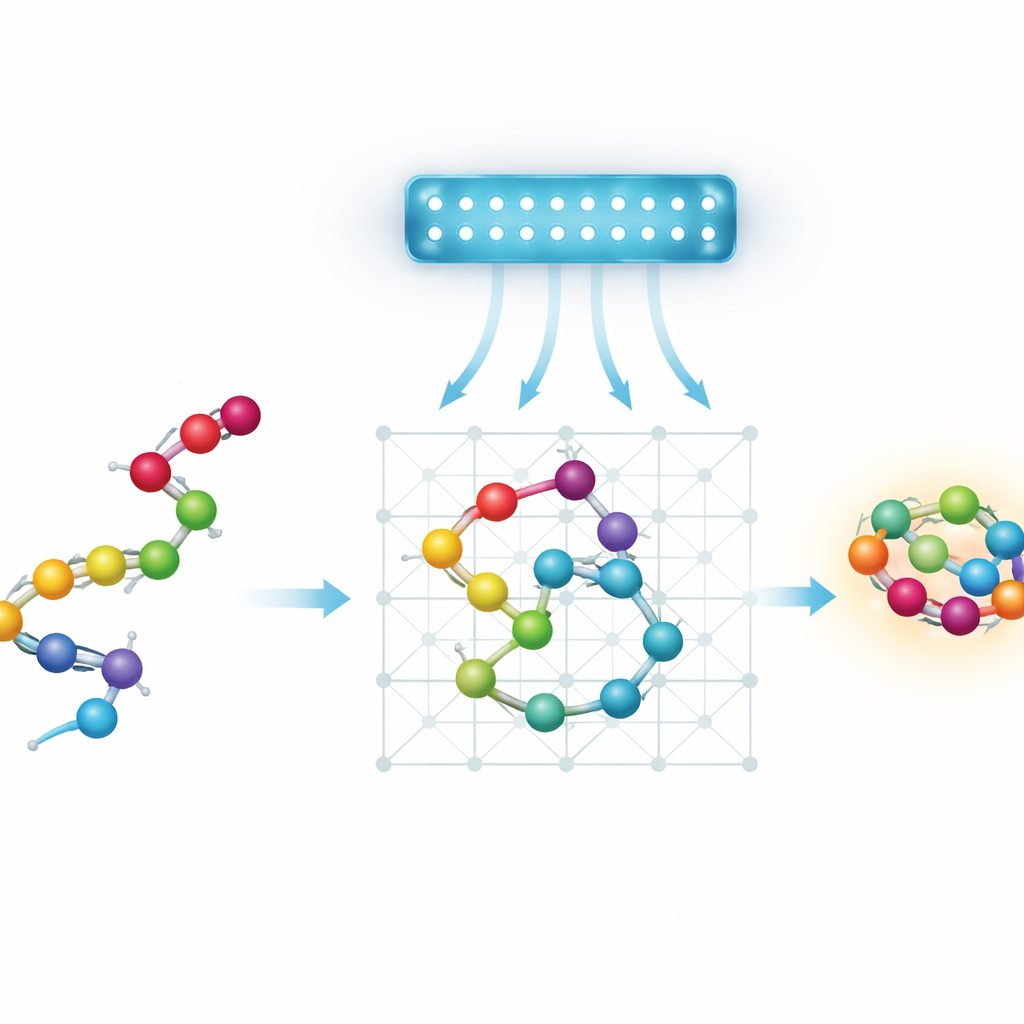
蛋白质、能量景观与寻找捷径
从本质上讲,蛋白质折叠是一个巨大的搜索问题。一条氨基酸链可以折叠成天文数量的形状,每种形状对应不同的能量。自然倾向于选择低能量构型,但“能量景观”通常崎岖不平,深谷之间由陡峭的山岭分隔。标准的优化方法和受物理启发的模拟常常陷入局部谷地,从而错过全局最优构型。量子退火具有吸引力,因为理论上量子效应(如隧穿)可能使系统穿过某些能垒,而不是总是爬过它们,从而有可能更快地找到良好的折叠状态。
为量子机器简化蛋白质
目前的量子退火器在规模和连通性上都有限,因此作者使用了简化的、或称粗粒化的蛋白质模型。与其追踪每个原子,他们将每个氨基酸表示为离散格点上的一个珠子。他们研究了四种将该设置编码为量子退火器可处理的数学问题的方法。两种“转弯式”(turn-based)模型通过链在格子上移动的步骤序列来描述;两种“坐标式”(coordinate-based)模型则将每个珠子分配到具体的晶格位置。每种变体可以基于常规的立方晶格或更经济的四面体晶格布局,后者每个珠子邻居更少。研究团队还提出了一种在四面体晶格上的新坐标式方案,使用两个交错的格子来表示交替的珠子,同时保持相互作用的简单性。
测试模型质量与问题难度
作者首先进行了一项详细的“资源审计”:随着蛋白质长度增加,每种模型需要多少二元变量(量子比特)、多少成对耦合,以及所需的相互作用强度范围有多宽。他们表明,某些转弯式编码会迅速变得过于密集并需要非常高的数值精度,这两点都与当前硬件不相容。一种先前提出的转弯式四面体模型暴露出更严重的问题:对于超过大约十个氨基酸的序列,它可能会将明显不符合物理的自交折叠视为最低能量的合法解。相比之下,坐标式方案天然只需要成对相互作用并使用更规则的惩罚项,这使得所需的数值精度保持适中,并且在映射到硬件时更为干净,尤其是在四面体晶格上。
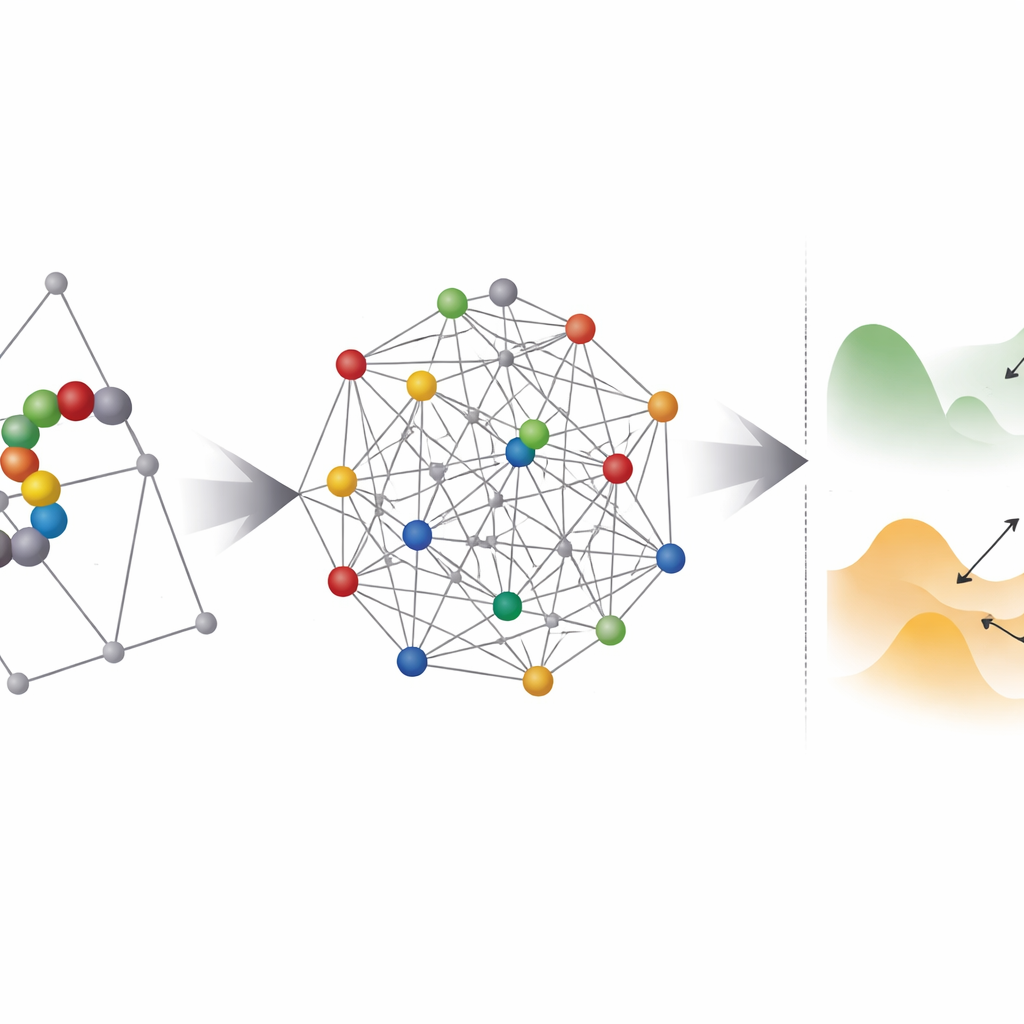
量子硬件能帮上忙的地方与它的局限
接着,团队研究了这些已编码的问题对任何优化器(量子或经典)而言在多大程度上是“崎岖”的。他们使用一种称为并行回火(parallel tempering)的技术来探测低能态的分布并估计它们之间能垒的厚度。大多数坐标式实例落入一种理论上量子隧穿可以帮助在相邻极小值之间跳跃的情形。然而,一个现实的障碍显现出来:现有量子退火器无法让每个量子比特与所有其他比特直接相连,因此每个逻辑变量必须通过一种称为嵌入的过程映射到一串物理量子比特上。这显著增加了所需量子比特数量,并且常常使有效的能量景观更加崎岖、更难求解。
量子与经典求解器的对决
最后,作者比较了不同方法找到短链随机氨基酸序列真实最低能量折叠所需的时间。他们针对最有前景的模型(四面体晶格上的新坐标式编码)对比了高度优化的 GPU 加速模拟退火代码和两代商用量子退火器。对于六到九残基的小测蛋白,两种方法随规模的扩展表现出相似的标度关系,但在绝对时间上,经典代码在原始问题上仍然快得多——快几个数量级。然而,当两种方法被要求求解完全相同、已嵌入的版本问题时,量子退火器的表现优于他们的经典模拟退火,这暗示一旦嵌入不再是主要瓶颈,量子方案可能在标度上具有潜在优势。
对未来道路的含义
对普通读者而言,结论是量子退火尚不是解决现实蛋白质折叠的捷径,但也不是死胡同。研究指出在稀疏四面体晶格上的坐标式模型是最有希望的路线,并表明模型设计强烈影响所得问题对任一求解器的难度。现有量子硬件在连通性和精度方面不足以处理超出概念验证规模的蛋白质,而且一些现有的折叠模型甚至对基本物理的描述存在误差。尽管如此,随着量子器件拥有更高连通性的量子比特和更低的误差率,以及编码方法经过改进以避免非物理折叠并减少嵌入开销,量子退火有可能变得具有竞争力——并且最终在那些特别崎岖、使经典和基于 AI 的方法吃力的蛋白质折叠难题上超越它们。
引用: Scheiber, T., Heller, M. & Giebel, A. Exploring quantum annealing for coarse-grained protein folding. Sci Rep 16, 12035 (2026). https://doi.org/10.1038/s41598-026-46916-w
关键词: 量子退火, 蛋白质折叠, 粗粒化模型, 优化算法, 量子计算硬件