Clear Sky Science · es
Explorando el annealing cuántico para el plegamiento de proteínas en modelos de grano grueso
Por qué importa para los medicamentos del futuro
Las formas en que las proteínas se pliegan determinan cómo funcionan en nuestros cuerpos, desde transportar oxígeno hasta reconocer virus. Predecir esas formas es vital para el diseño de fármacos, pero hacerlo con precisión para todas las proteínas sigue siendo extraordinariamente difícil. Este artículo plantea si un nuevo tipo de hardware informático—los anealadores cuánticos—podría algún día ayudar a resolver problemas de plegamiento de proteínas especialmente difíciles que desafían a la inteligencia artificial y a los superordenadores clásicos actuales.
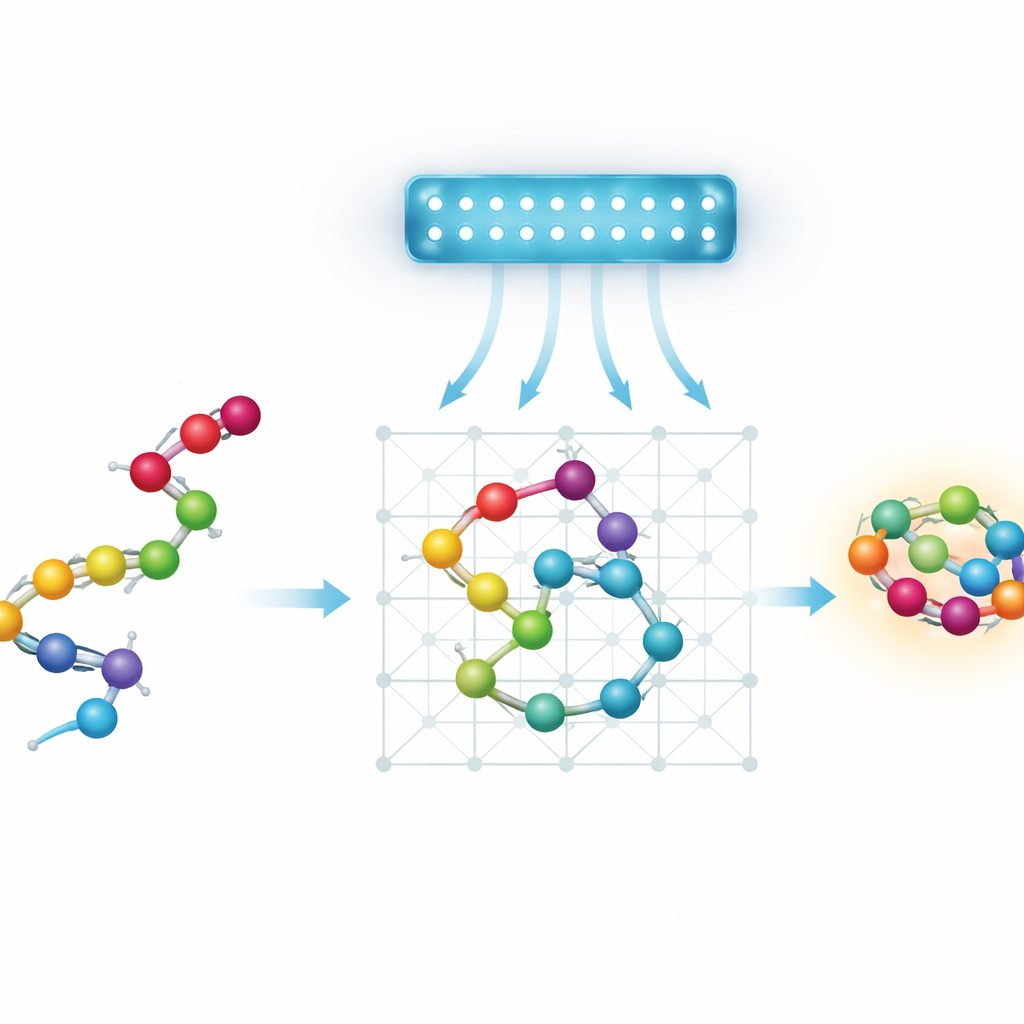
Proteínas, paisajes y la búsqueda de atajos
En el fondo, el plegamiento de proteínas es un problema de búsqueda gigantesco. Una cadena de aminoácidos puede plegarse en un número astronómico de formas, cada una con su propia energía. La naturaleza tiende a elegir las formas de baja energía, pero el “paisaje energético” es escarpado, lleno de valles profundos separados por colinas empinadas. Los métodos de optimización estándar y las simulaciones inspiradas en la física a menudo quedan atrapados en valles locales y pasan por alto la mejor conformación. El annealing cuántico resulta atractivo porque, en teoría, efectos cuánticos como el tunelamiento podrían permitir que un sistema atraviese algunas de estas colinas en lugar de tener que subirlas, encontrando potencialmente buenas conformaciones más rápido.
Simplificar proteínas para máquinas cuánticas
Los anealadores cuánticos actuales están limitados en tamaño y conectividad, por lo que los autores trabajan con modelos simplificados, o de grano grueso, de proteínas. En lugar de seguir cada átomo, representan cada aminoácido como una cuenta en una rejilla discreta. Examinaron cuatro maneras de codificar este planteamiento como un rompecabezas matemático que un anealador cuántico pueda abordar. Dos modelos “basados en giros” describen la cadena por la secuencia de pasos que realiza en una rejilla. Dos modelos “basados en coordenadas” asignan cada cuenta a una posición específica de la red. Cada variante puede disponerse sobre una rejilla cúbica estándar o sobre una rejilla tetraédrica más económica, donde cada cuenta tiene menos vecinas. El equipo también introduce un nuevo esquema basado en coordenadas en la red tetraédrica, usando dos rejillas entrelazadas para representar cuentas alternas mientras mantienen simples las interacciones.
Evaluar la calidad del modelo y la dureza del problema
Los autores realizan primero una “auditoría de recursos” detallada: cuántas variables binarias (qubits), cuántos acoplamientos pareados y qué rango de intensidades de interacción requiere cada modelo conforme crece la longitud de la proteína. Muestran que algunos codificadores basados en giros se vuelven rápidamente demasiado densos y requieren una precisión numérica muy alta, lo cual es incompatible con el hardware actual. Un modelo tetraédrico basado en giros propuesto anteriormente muestra un defecto más profundo: para secuencias de más de diez aminoácidos, puede considerar válidas conformaciones claramente no físicas con autointersecciones como soluciones de mínima energía. En contraste, los esquemas basados en coordenadas están naturalmente limitados a interacciones pareadas y usan penalizaciones más regulares, lo que mantiene la precisión numérica requerida en niveles modestos y hace más limpia la asignación al hardware, especialmente en la rejilla tetraédrica.
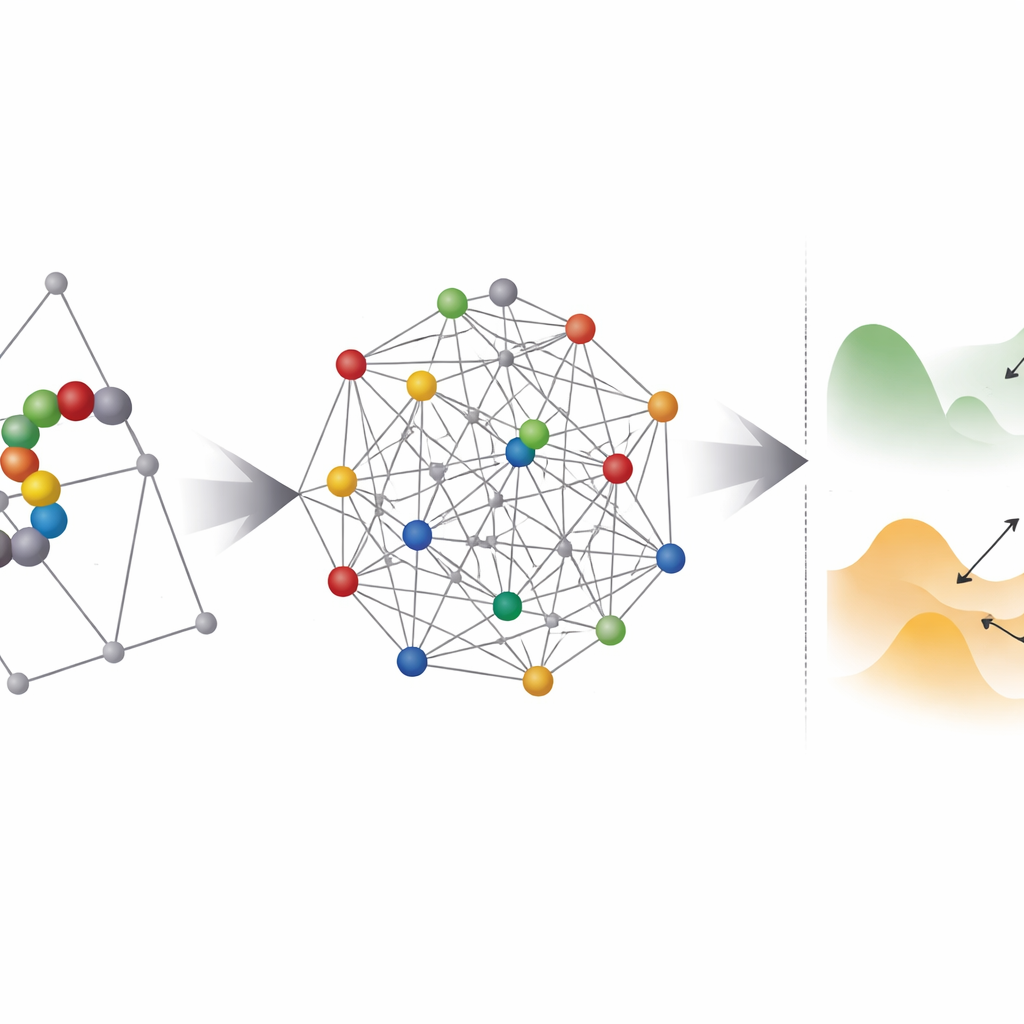
Dónde ayuda el hardware cuántico—y dónde flaquea
A continuación, el equipo investiga cuán “escabrosos” parecen estos problemas codificados para cualquier optimizador, cuántico o clásico. Usan una técnica llamada temple paralelo (parallel tempering) para sondear la distribución de estados de baja energía y estimar cuán gruesas son las barreras entre ellos. La mayoría de las instancias basadas en coordenadas caen en un régimen donde, en principio, el tunelamiento cuántico podría ayudar a saltar entre mínimos cercanos. Sin embargo, surge un obstáculo práctico: los anealadores cuánticos existentes no pueden conectar cada qubit con todos los demás, por lo que cada variable lógica debe mapearse a una cadena de qubits físicos en un proceso llamado embedding. Esto infla dramáticamente el número de qubits y a menudo hace que el paisaje energético efectivo sea más rugoso y más difícil de resolver.
Duelo entre solucionadores cuánticos y clásicos
Finalmente, los autores comparan cuánto tiempo tardan distintos métodos en encontrar la verdadera conformación de mínima energía para cadenas cortas de aminoácidos elegidas al azar. Miden el rendimiento de un código de temple simulado altamente optimizado y acelerado por GPU frente a dos generaciones de anealadores cuánticos comerciales, centrándose en el modelo más prometedor: la nueva codificación basada en coordenadas sobre la red tetraédrica. Para proteínas de prueba pequeñas de seis a nueve residuos, ambos enfoques muestran una escalada similar con el tamaño, pero en una escala absoluta el código clásico sigue siendo mucho más rápido—por varios órdenes de magnitud—cuando se ejecuta sobre el problema original. Sin embargo, cuando se pide a ambos métodos resolver exactamente la misma versión ya embebida del problema, el anealador cuántico rinde mejor que su temple simulado clásico, lo que insinúa una posible ventaja de escalado una vez que el embedding deje de ser el principal cuello de botella.
Qué significa esto para el camino por delante
Para un lector general, la conclusión es que el annealing cuántico todavía no es un atajo hacia plegar proteínas realistas, pero tampoco es un callejón sin salida. El estudio identifica los modelos basados en coordenadas sobre redes tetraédricas dispersas como la vía más prometedora, y muestra que el diseño del modelo moldea fuertemente la dificultad que plantean los problemas para cualquier solucionador. El hardware cuántico actual carece de la conectividad y la precisión para manejar proteínas mucho más allá de tamaños demostrativos, y algunos modelos de plegamiento existentes incluso describen mal física básica. Aun así, a medida que los dispositivos cuánticos incorporen qubits mejor conectados y tasas de error más bajas, y que se refinen las codificaciones para evitar pliegues no físicos y reducir la sobrecarga del embedding, el annealing cuántico podría volverse competitivo—y quizá eventualmente superior—para desafíos de plegamiento de proteínas especialmente escarpados que agotan a los métodos clásicos y basados en IA.
Cita: Scheiber, T., Heller, M. & Giebel, A. Exploring quantum annealing for coarse-grained protein folding. Sci Rep 16, 12035 (2026). https://doi.org/10.1038/s41598-026-46916-w
Palabras clave: annealing cuántico, plegamiento de proteínas, modelos de grano grueso, algoritmos de optimización, hardware de computación cuántica