Clear Sky Science · pt
Explorando o annealing quântico para o dobramento de proteínas em modelo grosseiro
Por que isso importa para medicamentos do futuro
As formas que as proteínas assumem determinam como elas funcionam em nossos corpos, desde transportar oxigênio até reconhecer vírus. Prever essas formas é vital para o desenvolvimento de fármacos, mas fazê-lo com precisão para todas as proteínas continua extremamente difícil. Este artigo pergunta se um novo tipo de hardware de computação — os annealers quânticos — poderia um dia ajudar a resolver problemas de dobramento de proteínas particularmente difíceis, que desafiam a inteligência artificial e os supercomputadores clássicos de hoje.
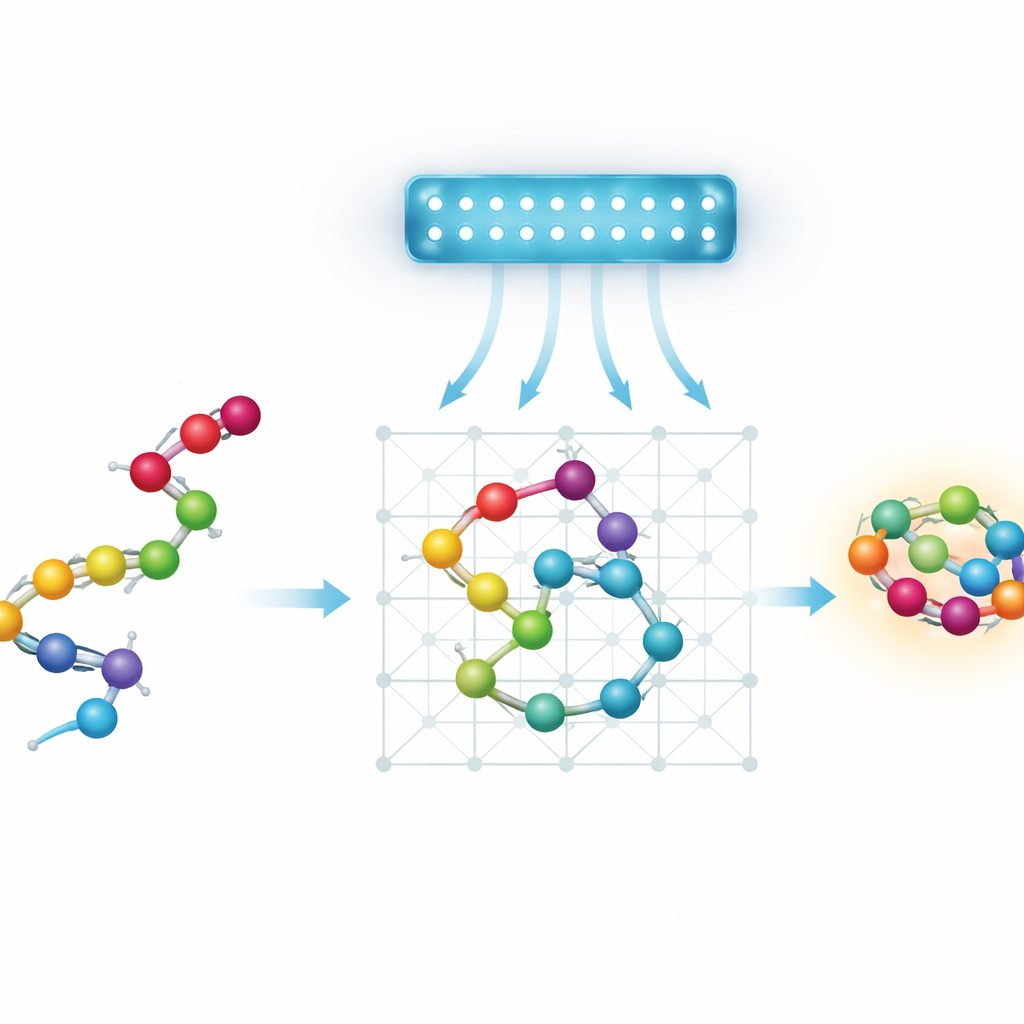
Proteínas, paisagens e uma busca por atalhos
No fundo, o dobramento de proteínas é um problema de busca gigantesco. Uma cadeia de aminoácidos pode dobrar em um número astronomicamente grande de formas, cada uma com sua própria energia. A natureza tende a escolher formas de baixa energia, mas a "paisagem de energia" é acidentada, cheia de vales profundos separados por colinas íngremes. Métodos padrão de otimização e simulações inspiradas na física frequentemente ficam presos em vales locais e perdem a melhor forma. O annealing quântico é atraente porque, em teoria, efeitos quânticos como tunelamento poderiam permitir que um sistema atravessasse algumas dessas colinas em vez de sempre escalá‑las, potencialmente encontrando bons dobramentos mais rápido.
Simplificando proteínas para máquinas quânticas
Os annealers quânticos atuais são limitados em tamanho e conectividade, então os autores trabalham com modelos simplificados, ou grosseiros, de proteínas. Em vez de acompanhar cada átomo, eles representam cada aminoácido como uma esfera em uma grade discreta. Eles examinam quatro maneiras de codificar essa configuração como um quebra-cabeça matemático que um annealer quântico pode resolver. Dois modelos "baseados em passos" descrevem a cadeia pela sequência de movimentos que ela faz em uma grade. Dois modelos "baseados em coordenadas" atribuem a cada esfera uma posição específica na rede. Cada variante pode ser montada em uma grade cúbica padrão ou em uma grade tetraédrica mais econômica, onde cada esfera tem menos vizinhos. A equipe também introduz um novo esquema baseado em coordenadas na rede tetraédrica, usando duas grades intercaladas para representar esferas alternadas enquanto mantém as interações simples.
Avaliando a qualidade do modelo e a dificuldade do problema
Os autores primeiro realizam uma "auditoria de recursos" detalhada: quantas variáveis binárias (qubits), quantos acoplamentos par a par e quão ampla é a faixa de forças de interação que cada modelo requer à medida que o comprimento da proteína cresce. Eles mostram que algumas codificações baseadas em passos tornam‑se rapidamente muito densas e exigem precisão numérica muito alta, ambos incompatíveis com o hardware atual. Um modelo tetraédrico baseado em passos proposto anteriormente revela um defeito mais profundo: para sequências com mais de cerca de dez aminoácidos, ele pode considerar dobras claramente não físicas, que se cruzam, como soluções legítimas de menor energia. Em contraste, os esquemas baseados em coordenadas são naturalmente limitados a interações par a par e usam penalidades mais regulares, o que mantém a precisão numérica requerida modesta e o mapeamento para o hardware mais limpo, especialmente na grade tetraédrica.
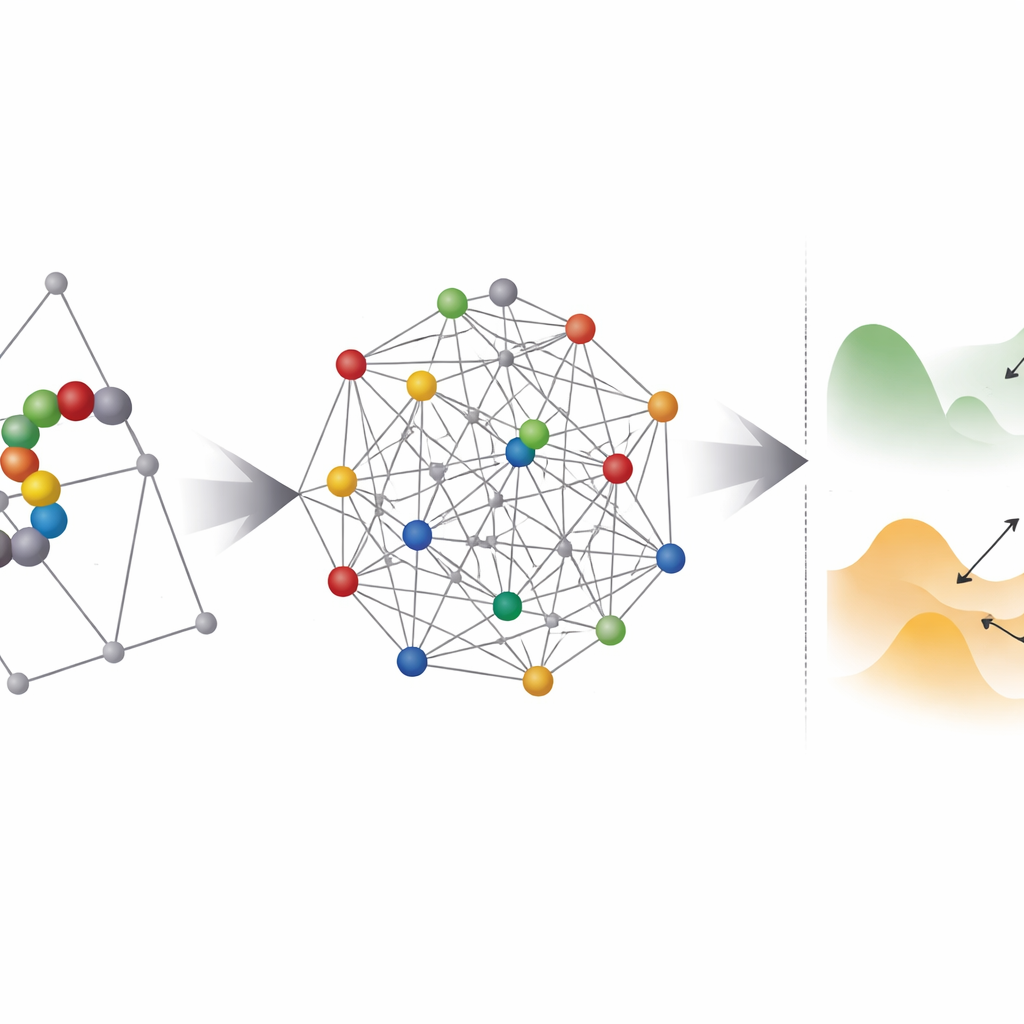
Onde o hardware quântico ajuda — e onde ele encontra dificuldades
Em seguida, a equipe investiga quão "acidentados" esses problemas codificados parecem para qualquer otimizador, quântico ou clássico. Eles usam uma técnica chamada temperamento paralelo para sondar a distribuição de estados de baixa energia e estimar quão espessas são as barreiras entre eles. A maioria das instâncias baseadas em coordenadas cai em um regime onde o tunelamento quântico poderia, em princípio, ajudar a saltar entre mínimos próximos. No entanto, surge um obstáculo prático: os annealers quânticos existentes não conseguem conectar todo qubit a todo outro, então cada variável lógica deve ser mapeada para uma cadeia de qubits físicos em um processo chamado embedding. Isso infla dramaticamente o número de qubits e frequentemente torna a paisagem de energia efetiva mais áspera e mais difícil de resolver.
Confronto entre solucionadores quânticos e clássicos
Finalmente, os autores comparam quanto tempo diferentes métodos levam para encontrar o verdadeiro dobramento de menor energia para cadeias curtas e aleatórias de aminoácidos. Eles comparam um código de annealing simulado altamente otimizado e acelerado por GPU contra duas gerações de annealers quânticos comerciais, focando no modelo mais promissor: a nova codificação baseada em coordenadas na rede tetraédrica. Para pequenas proteínas‑teste de seis a nove resíduos, ambas as abordagens mostram escalonamento semelhante com o tamanho, mas em termos absolutos o código clássico ainda é muito mais rápido — por várias ordens de grandeza — quando executado no problema original. No entanto, quando ambos os métodos são solicitados a resolver exatamente a mesma versão já embedded do problema, o annealer quântico tem desempenho superior ao annealing simulado clássico deles, sugerindo uma possível vantagem de escalabilidade quando o embedding deixar de ser o principal gargalo.
O que isso significa para o caminho à frente
Para um leitor geral, a conclusão é que o annealing quântico ainda não é um atalho para dobrar proteínas realistas, mas também não é um beco sem saída. O estudo identifica modelos baseados em coordenadas em malhas tetraédricas esparsas como o caminho mais promissor, e mostra que o desenho do modelo molda fortemente quão difíceis os problemas resultantes são para qualquer solucionador. O hardware quântico atual carece de conectividade e precisão para lidar com proteínas muito além de tamanhos de prova de conceito, e alguns modelos de dobramento existentes chegam a descrever mal física básica. Ainda assim, à medida que os dispositivos quânticos adicionarem qubits melhor conectados e taxas de erro mais baixas, e enquanto as codificações forem refinadas para evitar dobras não físicas e reduzir o overhead de embedding, o annealing quântico pode tornar‑se competitivo — e talvez eventualmente superior — para desafios de dobramento de proteínas especialmente acidentados que sobrecarregam métodos clássicos e baseados em IA.
Citação: Scheiber, T., Heller, M. & Giebel, A. Exploring quantum annealing for coarse-grained protein folding. Sci Rep 16, 12035 (2026). https://doi.org/10.1038/s41598-026-46916-w
Palavras-chave: annealing quântico, dobramento de proteínas, modelos grosseiros, algoritmos de otimização, hardware de computação quântica