Clear Sky Science · en
Exploring quantum annealing for coarse-grained protein folding
Why this matters for future medicines
The shapes that proteins fold into control how they work in our bodies, from carrying oxygen to recognizing viruses. Predicting these shapes is vital for drug design, but doing so accurately for all proteins is still extremely hard. This article asks whether a new kind of computing hardware—quantum annealers—could one day help crack especially tough protein-folding problems that challenge today’s artificial intelligence and classical supercomputers.
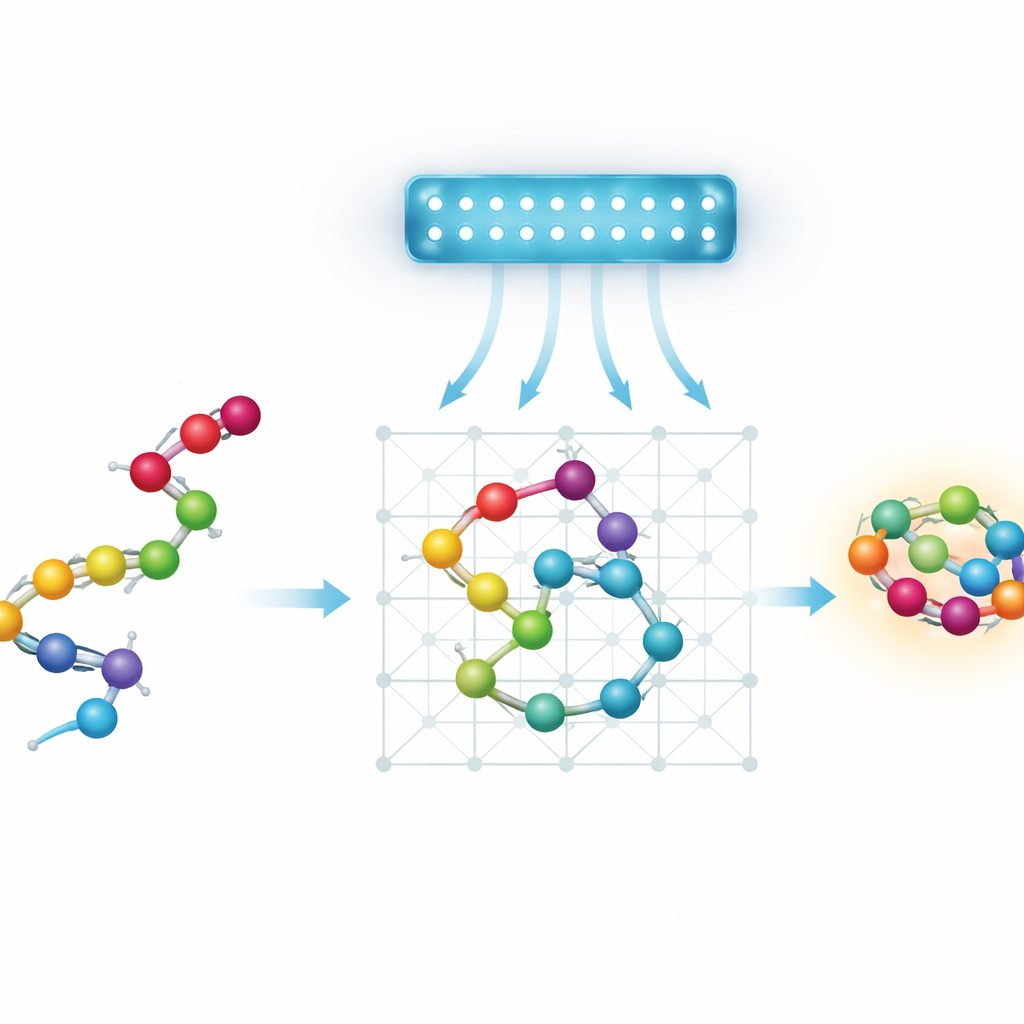
Proteins, landscapes, and a search for shortcuts
At heart, protein folding is a gigantic search problem. A chain of amino acids can fold into astronomically many shapes, each with its own energy. Nature tends to pick low-energy shapes, but the “energy landscape” is rugged, full of deep valleys separated by steep hills. Standard optimization methods and physics-inspired simulations often get trapped in local valleys and miss the best shape. Quantum annealing is attractive because, in theory, quantum effects such as tunneling could let a system slip through some of these hills instead of always climbing over them, potentially finding good folds faster.
Simplifying proteins for quantum machines
Today’s quantum annealers are limited in size and connectivity, so the authors work with simplified, or coarse-grained, models of proteins. Instead of tracking every atom, they represent each amino acid as a bead on a discrete grid. They examine four ways to encode this setup as a mathematical puzzle a quantum annealer can handle. Two “turn-based” models describe the chain by the sequence of steps it takes on a grid. Two “coordinate-based” models assign each bead to a specific lattice position. Each variant can be laid out on either a standard cubic grid or a more economical tetrahedral grid, where each bead has fewer neighbors. The team also introduces a new coordinate-based scheme on the tetrahedral lattice, using two interleaved grids to represent alternating beads while keeping interactions simple.
Testing model quality and problem hardness
The authors first perform a detailed “resource audit”: how many binary variables (qubits), how many pairwise couplings, and how wide a range of interaction strengths each model requires as protein length grows. They show that some turn-based encodings quickly become too dense and require very high numerical precision, both of which are bad matches for current hardware. One previously proposed turn-based tetrahedral model shows a deeper flaw: for sequences longer than about ten amino acids it can consider clearly unphysical self-crossing folds as legitimate lowest-energy solutions. In contrast, the coordinate-based schemes are naturally limited to pairwise interactions and use more regular penalties, which keeps the required numeric precision modest and the mapping to hardware cleaner, especially on the tetrahedral grid.
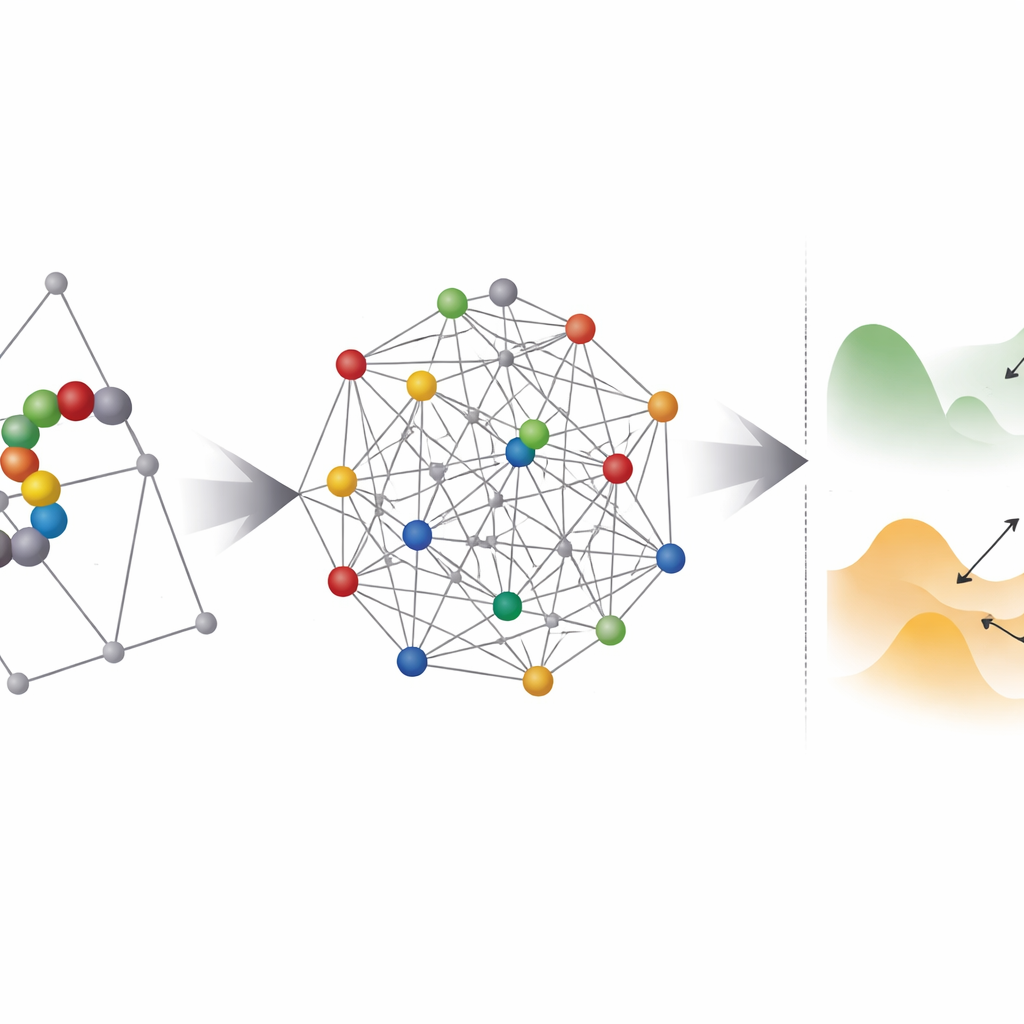
Where quantum hardware helps—and where it struggles
Next, the team investigates how “rugged” these encoded problems look to any optimizer, quantum or classical. They use a technique called parallel tempering to probe the distribution of low-energy states and estimate how thick the barriers are between them. Most of the coordinate-based instances fall into a regime where quantum tunneling could, in principle, help jump between nearby minima. However, a practical hurdle looms: existing quantum annealers cannot connect every qubit to every other, so each logical variable must be mapped onto a chain of physical qubits in a process called embedding. This inflates the number of qubits dramatically and often makes the effective energy landscape rougher and harder to solve.
Showdown between quantum and classical solvers
Finally, the authors compare how long different methods take to find the true lowest-energy fold for short, randomly chosen amino-acid chains. They benchmark a highly optimized, GPU-accelerated simulated annealing code against two generations of commercial quantum annealers, focusing on the most promising model: the new coordinate-based encoding on the tetrahedral grid. For small test proteins of six to nine residues, both approaches show similar scaling with size, but on an absolute scale the classical code is still much faster—by several orders of magnitude—when run on the original problem. However, when both methods are asked to solve exactly the same, already-embedded version of the problem, the quantum annealer performs better than their classical simulated annealing, hinting at a potential scaling edge once embedding is no longer the main bottleneck.
What this means for the road ahead
For a general reader, the takeaway is that quantum annealing is not yet a shortcut to folding realistic proteins, but it is not a dead end either. The study identifies coordinate-based models on sparse tetrahedral lattices as the most promising path forward, and it shows that model design strongly shapes how hard the resulting problems are for any solver. Today’s quantum hardware lacks the connectivity and precision to handle proteins far beyond proof-of-concept sizes, and some existing folding models even misdescribe basic physics. Still, as quantum devices add better-connected qubits and lower error rates, and as encodings are refined to avoid unphysical folds and reduce embedding overhead, quantum annealing could become competitive—and perhaps eventually superior—for especially rugged protein-folding challenges that strain classical and AI-based methods.
Citation: Scheiber, T., Heller, M. & Giebel, A. Exploring quantum annealing for coarse-grained protein folding. Sci Rep 16, 12035 (2026). https://doi.org/10.1038/s41598-026-46916-w
Keywords: quantum annealing, protein folding, coarse-grained models, optimization algorithms, quantum computing hardware