Clear Sky Science · zh
用于校正批次效应与整合多组学数据的统一框架
为何混合多种数据类型至关重要
现代生物学可以测量细胞中多种分子,从哪些基因处于活跃状态,到 DNA 的化学标记以及染色质的紧密程度。将这些层次一起研究——称为“多组学”——能够揭示诸如癌症或大脑连接模式等疾病中隐藏的规律。但由于这些测量成本高昂,研究者常常需要整合来自不同医院和实验室的数据,这会引入不希望出现的技术差异,模糊甚至掩盖真实的生物学信号。本文提出了 MoDAmix,一种新的计算方法,用于清理并合并此类复杂数据集,使研究人员能够信任所见结果。

凌乱的数据批次问题
当世界各地的实验室测量相似样本时,很少会使用完全相同的设备、方案或时间安排。这些差异会产生“批次效应”——由技术而非生物学引起的系统性偏差。在单组学研究中,已有若干工具尝试去除这些伪像,但它们各自独立处理每种测量类型。对于多组学数据,这种做法并不充分。如果对每一层分别清理,相同患者或细胞的不同分子视角可能会彼此偏离,破坏那些最具信息量的联系。作者认为,需要一种协调的方法,同时清理所有层并保持它们的共享结构完整。
一种对齐多层数据的新方法
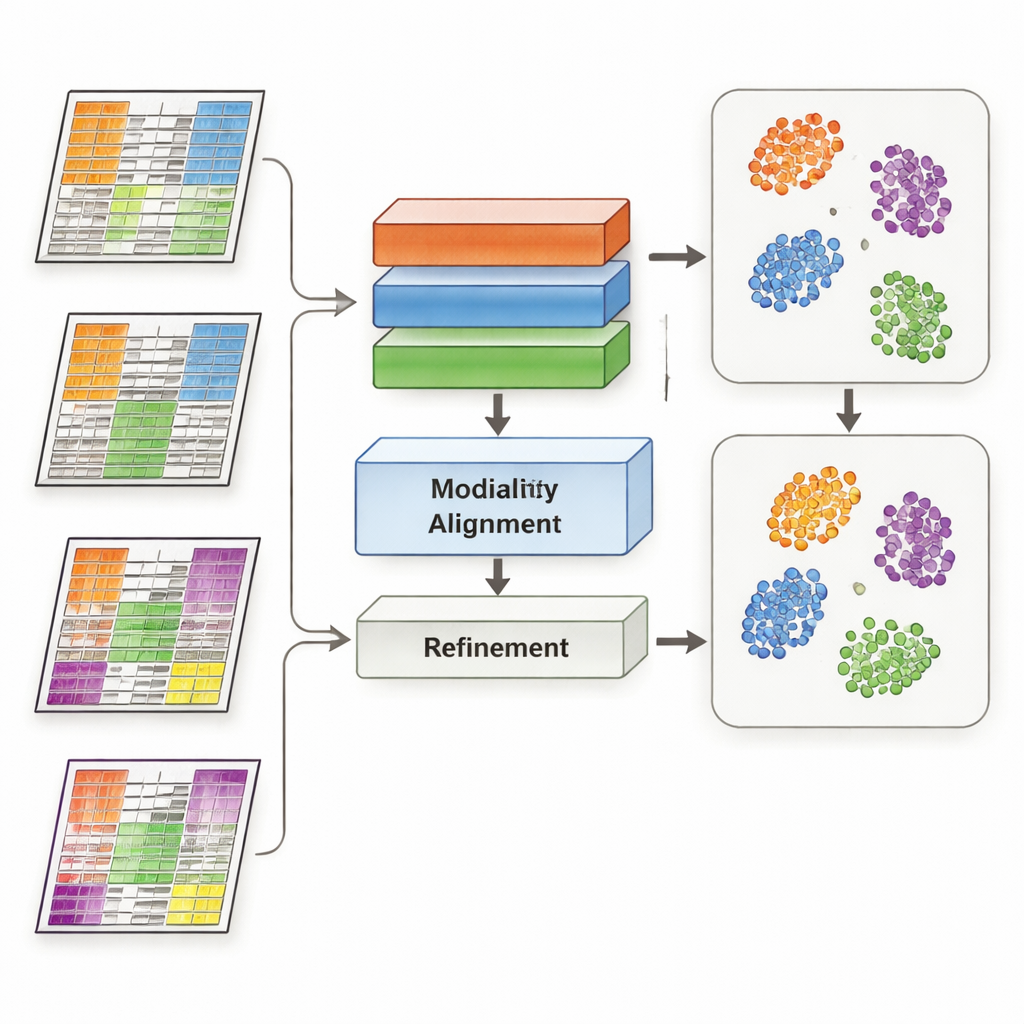
MoDAmix 通过借鉴“领域自适应”的思想来应对这一挑战,这是一种用于使模型在不同环境下也能工作的机器学习策略,例如处理来自不同相机的图像。该方法分四步进行。首先,它在有标签的“源”数据集中学习如何表示每种组学类型以及如何区分生物学亚型。第二,它通过训练模型使来自不同研究的样本在代表相同生物学时看起来相似,从而减少每个组学层内的批次效应。第三,它将所有组学类型汇聚到一个共享的低维空间,并再次强制模型忽略数据集间的技术差异。最后,它通过让模型对无标签的“目标”数据进行标签预测,并将同一亚型的样本在该共享空间中温和地拉向共同中心,以此来强化亚型边界。
将方法付诸检验
研究人员在三项具有挑战性的任务上测试了 MoDAmix。在一项成年小鼠大脑的单细胞研究中,他们结合了基因活性与染色质可及性来识别细胞类型。在两项癌症研究中,他们整合了基因表达与 DNA 甲基化,以跨独立患者队列对急性髓系白血病和脑肿瘤的亚型进行分类。他们将 MoDAmix 与流行的批次校正工具及较新的多组学整合方法进行了比较。使用分类准确率和聚类质量等衡量指标,MoDAmix 一贯产生更干净的细胞与患者分组。数据的可视化图显示,不同批次的样本混合良好,而不同细胞类型或肿瘤亚型仍清晰分离——这是竞争方法常常无法实现的。
为何对抗性学习有助于整合
MoDAmix 的一个关键成分是对抗性学习,其中模型的一部分尝试区分批次,而另一部分则学习使它们不可区分。为测试其重要性,作者移除了这些对抗模块并观察性能变化。去掉它们后,亚型预测的准确性下降,共享空间中的聚类变得更模糊且重叠增多。当仅在单组学层面保留对抗性对齐但不在联合多组学空间中实施时,结果有所改善但仍落后于完整模型。这些实验表明,在个体层面和联合层面上积极推动模型忽略批次特异信号,对于稳健的整合至关重要。
对未来研究的意义
MoDAmix 提供了一套通用方案,用于清理并统一来自多源的多组学数据,使得在脑细胞多样性、癌症亚型等领域更容易发现可靠的模式。通过谨慎地区分技术噪声与真实的生物差异,它使研究者能够在不丢失关键细微信号的情况下汇总不同队列的信息,从而有助于诊断、预后或治疗选择。随着多组学项目的不断扩展,像 MoDAmix 这样的工具可能会成为将庞大而凌乱的数据集转化为清晰、可操作见解的必备利器。
引用: Choi, J., Chae, H. A unified framework for correcting batch effects and integrating multi-omics data. Sci Rep 16, 12341 (2026). https://doi.org/10.1038/s41598-026-42355-9
关键词: 多组学整合, 批次效应校正, 癌症亚型划分, 单细胞分析, 领域自适应