Clear Sky Science · nl
Een eenduidig kader voor het corrigeren van batch-effecten en het integreren van multi-omics gegevens
Waarom het mengen van veel datatypes ertoe doet
De moderne biologie kan veel soorten moleculen in onze cellen meten, van welke genen actief zijn tot hoe DNA chemisch wordt gemarkeerd en hoe strak het is opgevouwen. Het gezamenlijk bestuderen van deze lagen, bekend als "multi-omics", kan verborgen patronen onthullen bij ziekten zoals kanker of in de bedrading van de hersenen. Omdat deze metingen echter duur zijn, combineren wetenschappers vaak gegevens uit veel ziekenhuizen en laboratoria, wat ongewenste technische verschillen introduceert die echte biologische signalen kunnen vervagen of verbergen. Dit artikel introduceert MoDAmix, een nieuwe computationele methode die dergelijke complexe datasets opschoont en samenvoegt, zodat onderzoekers kunnen vertrouwen op wat ze zien.

Het probleem van rommelige databatches
Wanneer laboratoria over de hele wereld vergelijkbare monsters meten, gebruiken ze zelden identieke apparaten, protocollen of timing. Deze verschillen veroorzaken "batch-effecten" – systematische eigenaardigheden veroorzaakt door technologie in plaats van biologie. In single-omics studies proberen verschillende tools deze artefacten al te verwijderen, maar ze behandelen elk type meting apart. Voor multi-omics data is dat niet voldoende. Als elke laag afzonderlijk wordt opgeschoond, kunnen de verschillende moleculaire gezichtspunten van dezelfde patiënt of cel uit elkaar drijven, waardoor de meest informatieve verbanden verbroken raken. De auteurs stellen dat wat nodig is een gecoördineerde aanpak die alle lagen samen opruimt terwijl hun gedeelde structuur behouden blijft.
Een nieuwe manier om veel datalagen op elkaar af te stemmen
MoDAmix pakt deze uitdaging aan door ideeën te lenen uit "domeinaanpassing", een machine-learningstrategie die wordt gebruikt om modellen te laten werken in verschillende omstandigheden, zoals afbeeldingen van verschillende camera's. De methode verloopt in vier stappen. Ten eerste leert het een representatie voor elk omics-type en hoe biologische subtypes te onderscheiden met een gelabelde "source"-dataset. Ten tweede vermindert het batch-effecten binnen elke omics-laag door het systeem te trainen zodat monsters uit verschillende studies vergelijkbaar lijken als ze dezelfde biologie vertegenwoordigen. Ten derde brengt het alle omics-typen samen in een gedeelde laag-dimensionale ruimte en dwingt het model opnieuw om technische verschillen tussen datasets te negeren. Ten slotte verscherpt het de subtype-grenzen door het model labels te laten raden voor de ongelabelde "target"-data en zachtjes monsters van hetzelfde subtype naar gemeenschappelijke centra in deze gedeelde ruimte te trekken.
De methode op de proef gesteld
De onderzoekers testten MoDAmix op drie veeleisende taken. In een single-cell studie van de volwassen muizenhersenen combineerden ze genactiviteit en chromatine-toegankelijkheid om celtypen te identificeren. In twee kankerstudies integreerden ze genexpressie met DNA-methylering om subtypes van acute myeloïde leukemie en hersentumoren te classificeren over onafhankelijke patiëntcohorten. Ze vergeleken MoDAmix met populaire batch-correctietools en met nieuwere multi-omics integratiemethoden. Met behulp van maatstaven zoals classificatienauwkeurigheid en clusteringkwaliteit produceerde MoDAmix consequent schonere groeperingen van cellen en patiënten. Visuele kaarten van de gegevens toonden dat monsters uit verschillende batches goed gemengd waren, terwijl verschillende celtypen of tumorsubtypes duidelijk gescheiden bleven – iets wat concurrerende methoden vaak niet wisten te bereiken.
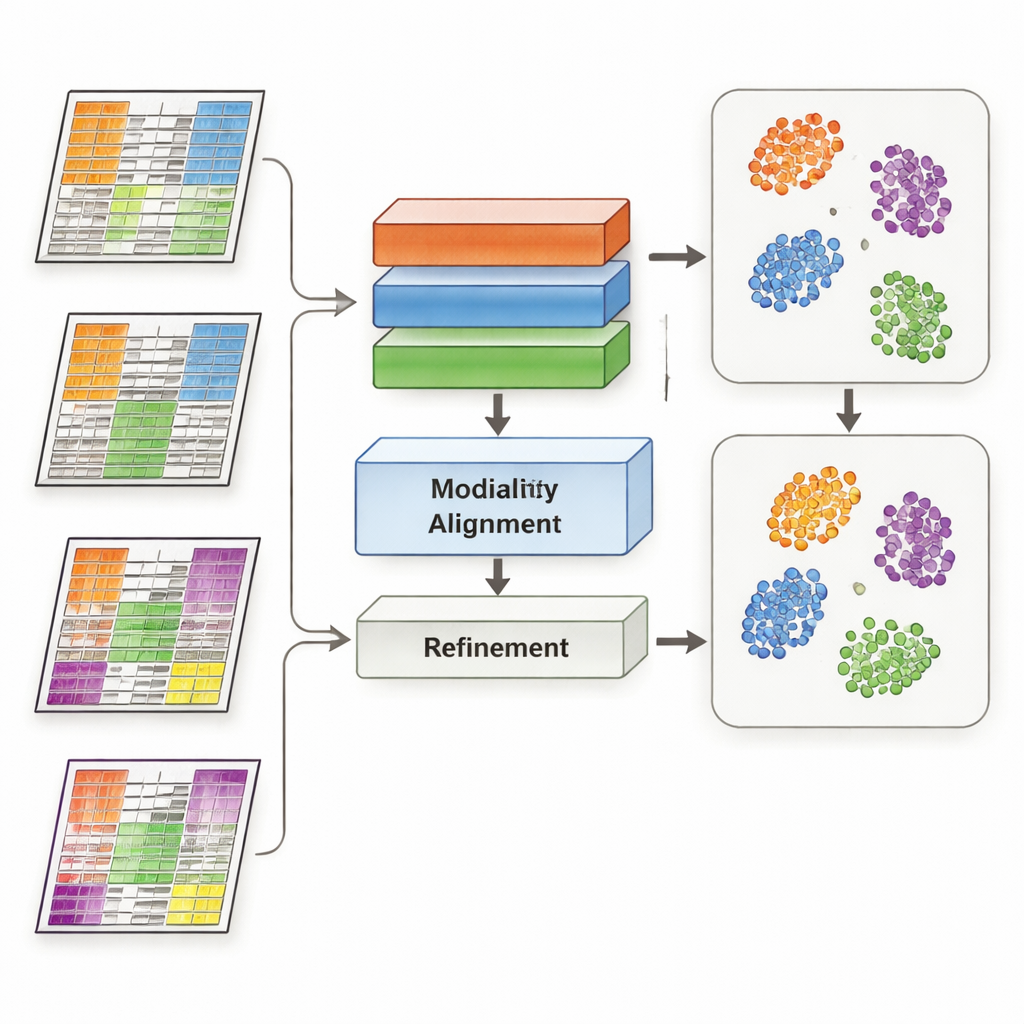
Waarom adversarial learning helpt
Een belangrijk ingrediënt in MoDAmix is adversarial learning, waarbij een deel van het model probeert batches van elkaar te onderscheiden terwijl een ander deel leert ze ononderscheidbaar te maken. Om het belang ervan te testen verwijderden de auteurs deze adversariële componenten en observeerden hoe de prestaties veranderden. Zonder deze componenten werden subtype-voorspellingen minder nauwkeurig en werden clusters in de gedeelde ruimte vager en meer overlappend. Wanneer adversariële afstemming alleen op single-omics niveau werd gehandhaafd maar niet in de gezamenlijke multi-omics ruimte, verbeterden de resultaten enigszins maar bleven ze achter bij het volledige model. Deze experimenten tonen aan dat het actief dwingen van het model om batch-specifieke signalen op zowel individueel als gecombineerd niveau te negeren cruciaal is voor robuuste integratie.
Wat dit betekent voor toekomstige studies
MoDAmix biedt een algemeen recept voor het opschonen en verenigen van multi-omics gegevens uit veel bronnen, waardoor het gemakkelijker wordt betrouwbare patronen te vinden in de diversiteit van hersencellen, kanker-subtypes en daarbuiten. Door technische ruis zorgvuldig te scheiden van echte biologische verschillen stelt het onderzoekers in staat informatie over cohorten heen te combineren zonder de subtiele signalen te verliezen die van belang zijn voor diagnose, prognose of behandelingskeuze. Naarmate multi-omics projecten blijven groeien, kunnen tools zoals MoDAmix essentieel worden om enorme, rommelige datasets om te zetten in heldere, toepasbare inzichten.
Bronvermelding: Choi, J., Chae, H. A unified framework for correcting batch effects and integrating multi-omics data. Sci Rep 16, 12341 (2026). https://doi.org/10.1038/s41598-026-42355-9
Trefwoorden: integratie van multi-omics, correctie van batch-effecten, kanker-subtyping, single-cell analyse, domeinaanpassing