Clear Sky Science · en
A unified framework for correcting batch effects and integrating multi-omics data
Why Mixing Many Data Types Matters
Modern biology can measure many kinds of molecules in our cells, from which genes are active to how DNA is chemically tagged and how tightly it is packed. Studying these layers together, known as “multi‑omics,” can reveal hidden patterns in diseases like cancer or in the wiring of the brain. But because these measurements are expensive, scientists often combine data from many hospitals and labs, which introduces unwanted technical differences that can blur or even hide true biological signals. This paper introduces MoDAmix, a new computational method that cleans and combines such complex datasets so that researchers can trust what they see.

The Problem of Messy Data Batches
When labs around the world measure similar samples, they rarely use identical machines, protocols, or timing. These differences create “batch effects” – systematic quirks caused by technology rather than biology. In single‑omics studies, several tools already try to remove these artifacts, but they treat each type of measurement alone. For multi‑omics data, this is not enough. If each layer is cleaned separately, the different molecular views of the same patient or cell can drift apart, breaking the links that are most informative. The authors argue that what is needed instead is a coordinated approach that cleans all layers together while keeping their shared structure intact.
A New Way to Align Many Data Layers
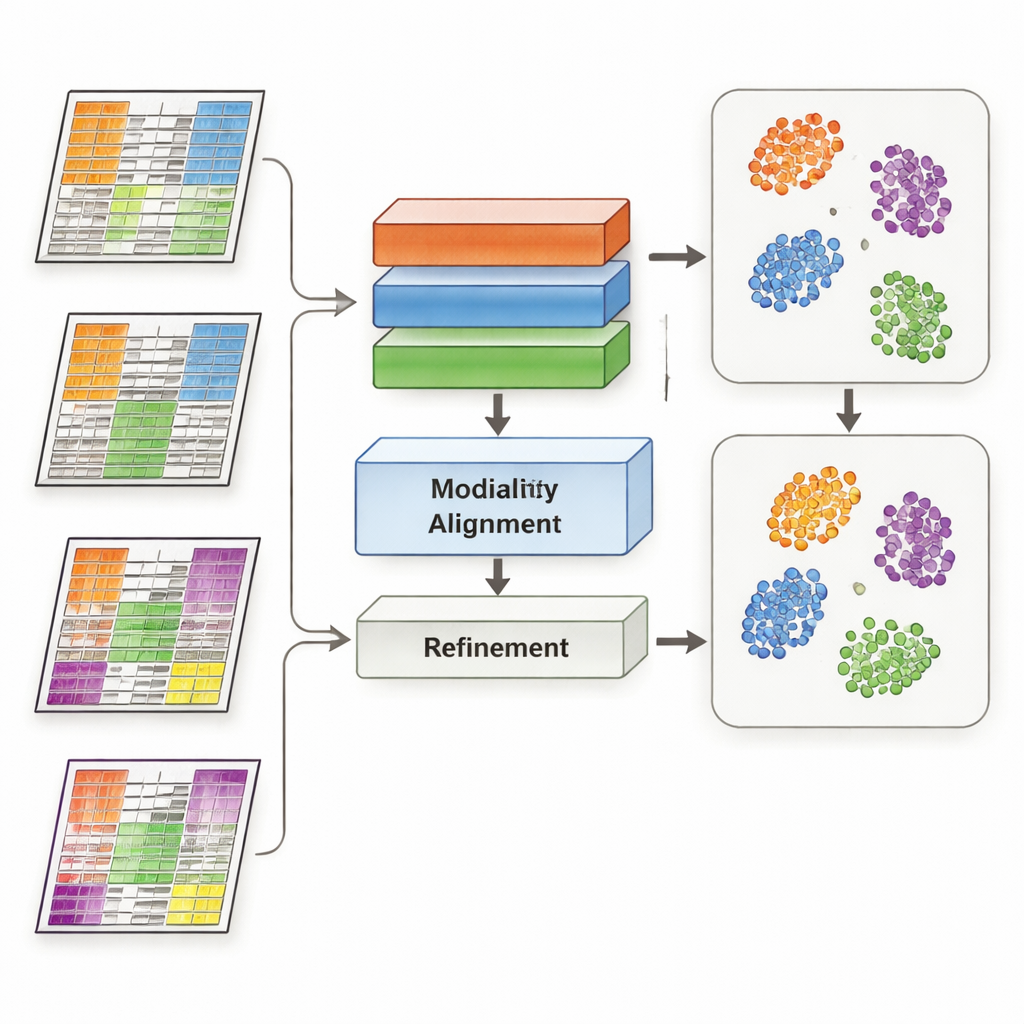
MoDAmix tackles this challenge by borrowing ideas from “domain adaptation,” a machine‑learning strategy used to make models work across different settings, such as images from different cameras. The method proceeds in four steps. First, it learns how to represent each omics type and how to distinguish biological subtypes using a labeled “source” dataset. Second, it reduces batch effects within each omics layer by training the system to make samples from different studies look similar if they represent the same biology. Third, it brings all omics types together into a shared low‑dimensional space and again forces the model to ignore technical differences between datasets. Finally, it sharpens subtype boundaries by letting the model guess labels for the unlabeled “target” data and gently pulling samples of the same subtype toward common centers in this shared space.
Putting the Method to the Test
The researchers tested MoDAmix on three demanding tasks. In a single‑cell study of adult mouse brain, they combined gene activity and chromatin accessibility to identify cell types. In two cancer studies, they integrated gene expression with DNA methylation to classify subtypes of acute myeloid leukemia and brain tumors across independent patient cohorts. They compared MoDAmix with popular batch‑correction tools and with newer multi‑omics integration methods. Using measures such as classification accuracy and clustering quality, MoDAmix consistently produced cleaner groupings of cells and patients. Visual maps of the data showed that samples from different batches were well mixed, while distinct cell types or tumor subtypes remained clearly separated – something competing methods often failed to achieve.
Why Adversarial Learning Helps
A key ingredient in MoDAmix is adversarial learning, in which one part of the model tries to tell batches apart while another part learns to make them indistinguishable. To test its importance, the authors removed these adversarial pieces and observed how performance changed. Without them, subtype predictions became less accurate, and clusters in the shared space became fuzzier and more overlapping. When adversarial alignment was kept only at the single‑omics level but not in the joint multi‑omics space, results improved somewhat but still lagged behind the full model. These experiments show that actively pushing the model to ignore batch‑specific signals at both individual and combined levels is crucial for robust integration.
What This Means for Future Studies
MoDAmix offers a general recipe for cleaning and unifying multi‑omics data from many sources, making it easier to find reliable patterns in brain cell diversity, cancer subtypes, and beyond. By carefully separating technical noise from genuine biological differences, it allows researchers to pool information across cohorts without losing the subtle signals that matter for diagnosis, prognosis, or treatment choice. As multi‑omics projects continue to grow, tools like MoDAmix could become essential for turning vast, messy datasets into clear, actionable insight.
Citation: Choi, J., Chae, H. A unified framework for correcting batch effects and integrating multi-omics data. Sci Rep 16, 12341 (2026). https://doi.org/10.1038/s41598-026-42355-9
Keywords: multi-omics integration, batch effect correction, cancer subtyping, single-cell analysis, domain adaptation