Clear Sky Science · de
Ein einheitliches Rahmenwerk zur Korrektur von Batch‑Effekten und Integration von Multi‑Omics‑Daten
Warum das Mischen vieler Datentypen wichtig ist
Die moderne Biologie kann viele Arten von Molekülen in unseren Zellen messen – von der aktiven Genexpression über chemische Markierungen der DNA bis hin zur Verpackungsdichte des Erbguts. Diese Ebenen gemeinsam zu untersuchen, als „Multi‑Omics“ bezeichnet, kann verborgene Muster bei Krankheiten wie Krebs oder in der Verschaltung des Gehirns aufdecken. Weil solche Messungen jedoch aufwendig sind, kombinieren Forschende häufig Daten aus vielen Kliniken und Laboren, was unerwünschte technische Unterschiede einbringt, die echte biologische Signale verwischen oder verbergen können. Dieses Papier stellt MoDAmix vor, eine neue Rechenmethode, die solche komplexen Datensätze bereinigt und zusammenführt, sodass Wissenschaftlerinnen und Wissenschaftler den Ergebnissen vertrauen können.

Das Problem unordentlicher Daten‑Batches
Wenn Labore weltweit ähnliche Proben messen, verwenden sie selten exakt dieselben Geräte, Protokolle oder Zeitpläne. Diese Unterschiede erzeugen „Batch‑Effekte“ – systematische Abweichungen, die von der Technologie statt von der Biologie stammen. In Einzel‑Omics‑Studien versuchen bereits mehrere Werkzeuge, solche Artefakte zu entfernen, doch sie behandeln jeden Messdatentyp isoliert. Für Multi‑Omics‑Daten ist das nicht ausreichend. Wenn jede Ebene separat bereinigt wird, können die verschiedenen molekularen Ansichten desselben Patienten oder derselben Zelle auseinanderdriften und die informativsten Verknüpfungen zerstören. Die Autoren argumentieren, dass stattdessen ein koordiniertes Vorgehen nötig ist, das alle Ebenen gemeinsam säubert und gleichzeitig ihre gemeinsame Struktur bewahrt.
Ein neuer Weg, viele Datenschichten auszurichten
MoDAmix begegnet dieser Herausforderung, indem es Ideen aus der „Domänenanpassung“ aufgreift, einer Machine‑Learning‑Strategie, die Modelle in unterschiedlichen Umgebungen – etwa Bildern verschiedener Kameras – funktionsfähig macht. Die Methode verläuft in vier Schritten. Zuerst lernt sie, jede Omics‑Art zu repräsentieren und biologische Subtypen anhand eines beschrifteten „Source“‑Datensatzes zu unterscheiden. Zweitens reduziert sie Batch‑Effekte innerhalb jeder Omics‑Ebene, indem das System so trainiert wird, dass Proben aus verschiedenen Studien ähnlich aussehen, wenn sie dieselbe Biologie repräsentieren. Drittens führt sie alle Omics‑Typen in einen gemeinsamen, niedrigdimensionalen Raum zusammen und zwingt das Modell erneut, technische Unterschiede zwischen Datensätzen zu ignorieren. Schließlich schärft sie die Subtyp‑Grenzen, indem das Modell Labels für die unbeschrifteten „Target“‑Daten schätzt und Proben desselben Subtyps behutsam in diesem gemeinsamen Raum zu gemeinsamen Zentren zieht.
Die Methode im Praxistest
Die Forschenden testeten MoDAmix an drei anspruchsvollen Aufgaben. In einer Einzelzellstudie des erwachsenen Mausgehirns kombinierten sie Genaktivität und Chromatinzugänglichkeit, um Zelltypen zu identifizieren. In zwei Krebsstudien integrierten sie Genexpressionsdaten mit DNA‑Methylierung, um Subtypen von akuter myeloischer Leukämie und Hirntumoren über unabhängige Patienten‑Kohorten hinweg zu klassifizieren. Sie verglichen MoDAmix mit gängigen Batch‑Korrekturwerkzeugen und neueren Multi‑Omics‑Integrationsmethoden. Mit Metriken wie Klassifikationsgenauigkeit und Clusterqualität erzeugte MoDAmix durchweg sauberere Gruppierungen von Zellen und Patienten. Visuelle Karten der Daten zeigten, dass Proben aus verschiedenen Batches gut durchmischt waren, während unterschiedliche Zelltypen oder Tumor‑Subtypen klar getrennt blieben – etwas, das konkurrierende Methoden oft nicht erreichten.
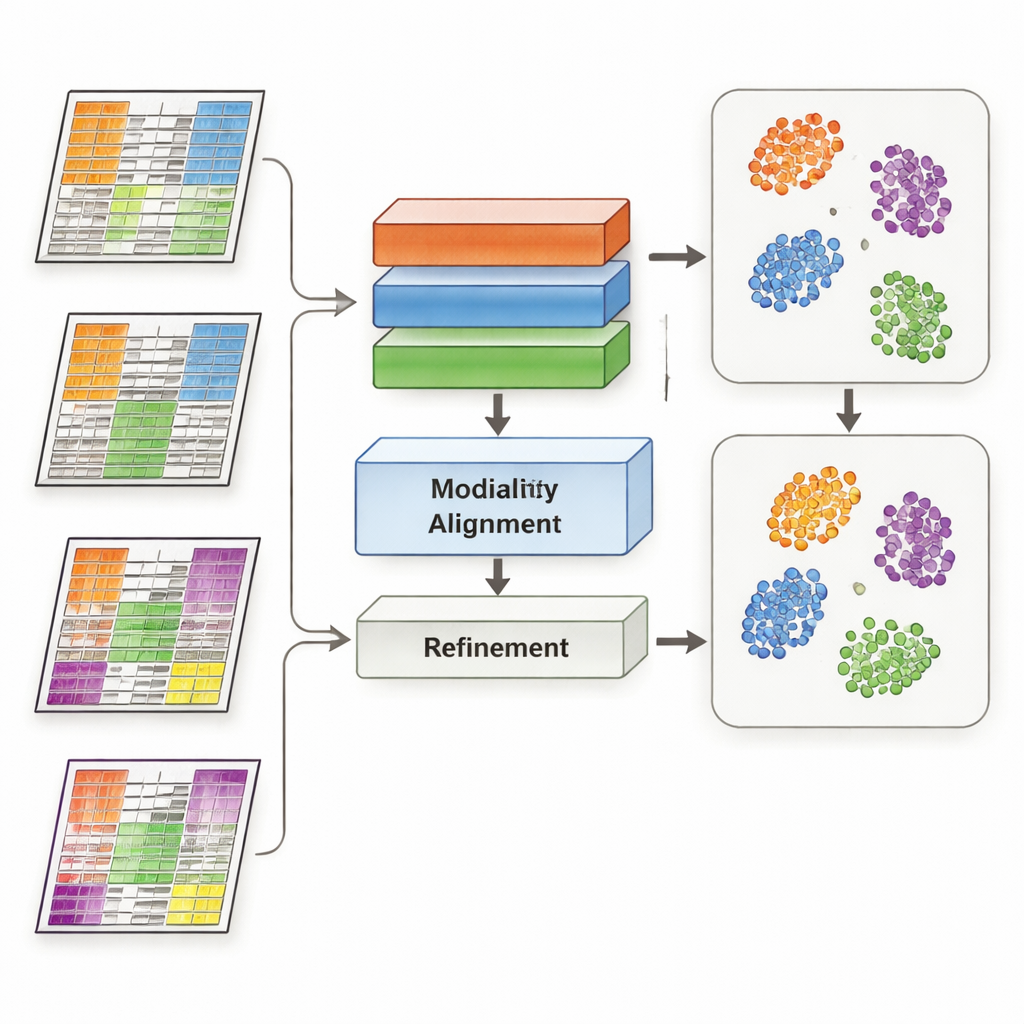
Warum adversariales Lernen hilft
Ein zentrales Element von MoDAmix ist adversariales Lernen, bei dem ein Teil des Modells versucht, Batches zu unterscheiden, während ein anderer Teil lernt, sie ununterscheidbar zu machen. Um dessen Bedeutung zu prüfen, entfernten die Autoren diese adversarialen Komponenten und beobachteten die Leistungsänderung. Ohne sie wurden Subtyp‑Vorhersagen weniger genau und die Cluster im gemeinsamen Raum wurden diffuser und überlappender. Wenn die adversariale Ausrichtung nur auf Einzel‑Omics‑Ebene beibehalten, nicht aber im gemeinsamen Multi‑Omics‑Raum angewendet wurde, verbesserten sich die Ergebnisse zwar etwas, blieben aber hinter dem vollständigen Modell zurück. Diese Experimente zeigen, dass es entscheidend ist, das Modell aktiv dazu zu bringen, batch‑spezifische Signale sowohl auf individueller als auch auf kombinierter Ebene zu ignorieren, um eine robuste Integration zu erreichen.
Was das für künftige Studien bedeutet
MoDAmix bietet ein allgemeines Rezept, um Multi‑Omics‑Daten aus vielen Quellen zu bereinigen und zu vereinigen, wodurch es einfacher wird, verlässliche Muster in der Vielfalt von Gehirnzellen, Krebs‑Subtypen und darüber hinaus zu finden. Indem technische Störungen sorgfältig von echten biologischen Unterschieden getrennt werden, ermöglicht es Forschenden, Informationen über Kohorten hinweg zu bündeln, ohne die feinen Signale zu verlieren, die für Diagnose, Prognose oder Therapieentscheidung wichtig sind. Mit dem weiteren Wachstum von Multi‑Omics‑Projekten könnten Werkzeuge wie MoDAmix entscheidend werden, um riesige, unordentliche Datensätze in klare, verwertbare Erkenntnisse zu verwandeln.
Zitation: Choi, J., Chae, H. A unified framework for correcting batch effects and integrating multi-omics data. Sci Rep 16, 12341 (2026). https://doi.org/10.1038/s41598-026-42355-9
Schlüsselwörter: Integration von Multi‑Omics, Korrektur von Batch‑Effekten, Krebs‑Subtypen, Einzelzell‑Analyse, Domänenanpassung