Clear Sky Science · zh
使用针对人类的反义寡核苷酸唤醒父源UBE3A的异种移植模型
为什么唤醒沉默基因很重要
安杰尔曼综合征是一种罕见但严重的脑部疾病,会导致儿童发育迟缓、癫痫发作和严重的言语障碍。该病源于神经细胞中一个关键基因UBE3A的丢失。不同寻常的是,每个神经元仍然携带来自父方的健康备份拷贝,但该拷贝通常处于关闭状态。本研究探索了一种使用精确基因药物“唤醒”这一沉默备份的方法,并引入了一种新的动物模型,使研究者能够在将这些针对人类定制的治疗用于患者之前,对其进行安全测试。
一种带有隐性备份的疾病
在身体大多数细胞中,UBE3A基因在母源和父源染色体上都处于活跃状态。然而在脑细胞中,仅有母源拷贝被开启;父源拷贝被一段沿DNA反方向延伸的长RNA所阻断,就像迎面而来的列车阻止其他交通通行那样。患有安杰尔曼综合征的儿童遗传了受损的母源UBE3A,导致其神经元缺乏功能性UBE3A蛋白,即便完整的父源拷贝存在。早期在小鼠中的研究显示,在生命早期恢复UBE3A可以挽救许多类似安杰尔曼的症状,这表明如果能恢复缺失的信号,大脑可能出人意料地具有一定的可逆性。
用短小的基因向导突破阻断
一种有前景的方法是使用反义寡核苷酸(ASO)来释放沉睡的父源基因——ASO是短小的人工合成核酸片段,能够与特定的RNA靶点结合。当针对阻断父源UBE3A的长阻断RNA设计ASO时,这些ASO可以促使该阻断RNA被降解,从而使父源基因重新表达。研究团队首先在来自安杰尔曼患者和健康志愿者的皮肤衍生干细胞分化形成的神经元中测试了一种针对人类的ASO。他们确认,随着细胞成熟为神经元,安杰尔曼细胞中的UBE3A逐渐消失,而健康细胞中则维持稳定。用针对阻断RNA的反义药物处理这些培养物后,阻断RNA显著减少,UBE3A的RNA水平提高,UBE3A蛋白恢复到相当接近正常的水平——且未见明显毒性。
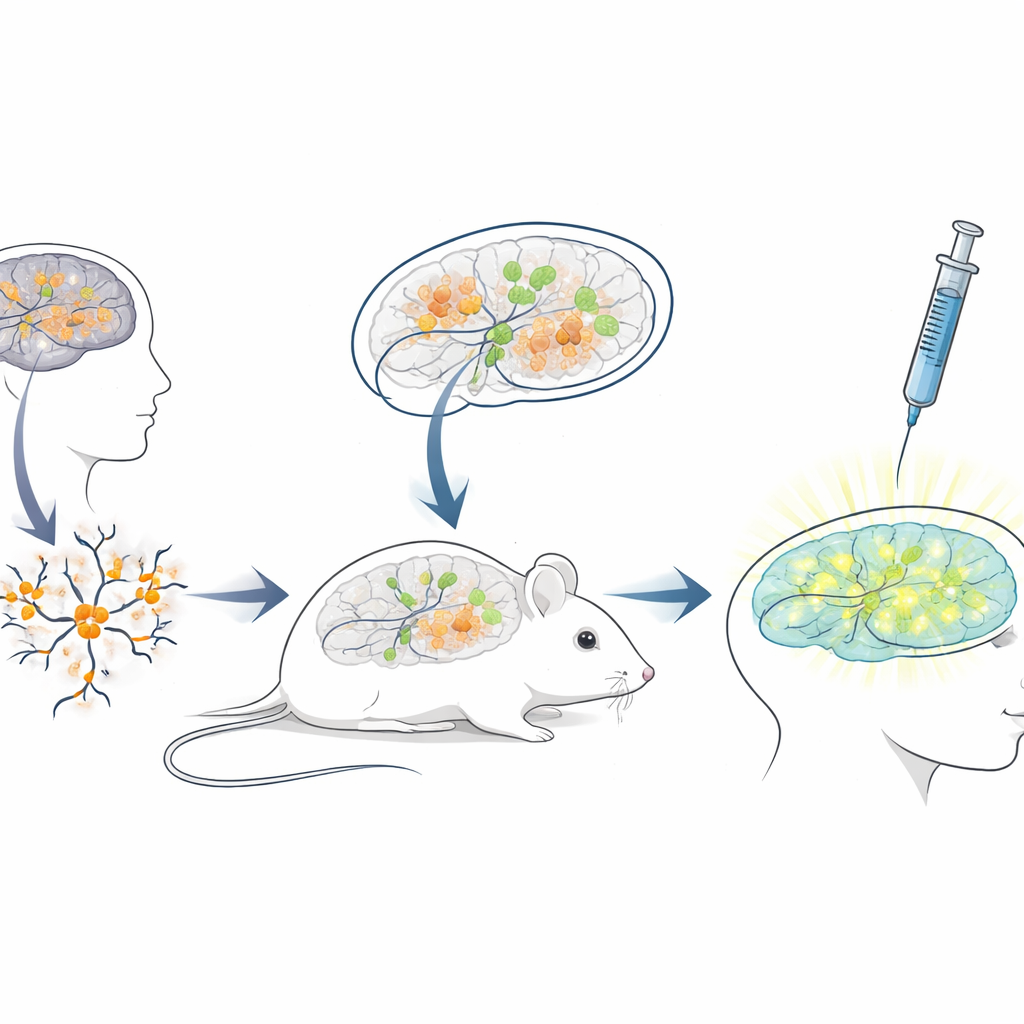
为人类疗法构建一个活体小鼠测试平台
由于人类的阻断RNA序列与小鼠差异很大,标准的小鼠模型不适合用于测试面向患者的人类ASO。为弥补这一差距,研究人员构建了一个“异种移植”平台:他们从携带安杰尔曼突变的人类干细胞中培养出神经元,然后将这些年轻的人类神经元移植到新生免疫缺陷小鼠的大脑中。移植的细胞定殖于海马等脑区,存活数月,并出现预期的、核内丧失UBE3A的安杰尔曼样表型。这创造了一种独特的情形:人类疾病神经元在小鼠大脑内生长和成熟,暴露于自然的脑回路以及临床上医生会使用的相同给药途径。
在更真实的环境中测试重新激活基因的药物
在这个人鼠嵌合大脑系统建立后,团队模拟了临床治疗流程。当小鼠三周大时,他们将反义药物注入脑内充满液体的腔隙,沿用了其他神经系统疾病已获批ASO药物的常用给药途径。一周后,脑切片显示,药物处理组中移植的人类神经元出现了明显的核内UBE3A信号,而只接受盐溶液的对照组则仍然暗淡。定量分析显示,近四分之三的移植人类安杰尔曼神经元恢复了可检测到的UBE3A,且平均信号接近健康对照神经元的重要水平。重要的是,小鼠对注射耐受良好,在治疗期间体重与对照组无差异。
这对未来精准药物的意义
这项工作尚不能治愈安杰尔曼综合征,但它提供了两项关键进展。首先,它表明一种针对人类的反义药物能够在患者来源的神经元中可靠地开启沉默的父源UBE3A——无论是在培养皿内,还是在这些神经元整合入活体大脑之后。其次,它提供了一个多用途的测试平台:携带真实患者突变的人类神经元可以被培养、移植到小鼠体内,并通过临床相关的途径暴露于候选疗法。随着罕见儿童脑病的基因诊断愈发普及,这类“个性化测试床”可能有助于评估为特定突变量身定制的反义治疗的安全性和有效性——即使全球仅有少数患者拥有相同的突变。
引用: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
关键词: 安杰尔曼综合征, 反义寡核苷酸, 异种移植, 基因重新激活, 神经发育障碍