Clear Sky Science · ru
Модель ксенотрансплантации для реактивации отцовского UBE3A с помощью антителоподобных олигонуклеотидов, специфичных для человека
Почему важно «разбудить» молчащий ген
Синдром Ангельмана — редкое, но тяжёлое заболевание мозга, вызывающее задержку развития, судороги и серьёзные проблемы с речью у детей. Оно возникает из‑за потери одного ключевого гена, называемого UBE3A, в нервных клетках. Необычно то, что в каждом нейроне остаётся здоровая запасная копия этого гена от отца, но эта копия обычно отключена. В этом исследовании изучают способ «разбудить» этот молчащий запасной ген с помощью точного генетического лекарства и представляют новую модель на животных, которая позволяет безопасно тестировать такие ориентированные на человека терапии перед их применением у пациентов.
Заболевание с скрытым запасным планом
В большинстве клеток организма ген UBE3A активен на хромосомах и матери, и отца. В мозговых клетках, однако, включена только материнская копия; отцовская блокируется длинной молекулой РНК, идущей в противоположном направлении по ДНК, словно поезд навстречу другому движению, мешающий проходу. Дети с синдромом Ангельмана наследуют повреждённую материнскую копию UBE3A, из‑за чего их нейроны остаются без работающего белка UBE3A, хотя неповреждённая отцовская копия присутствует. Ранние исследования на мышах показали, что восстановление UBE3A в раннем возрасте может исправить многие симптомы, похожие на проявления синдрома Ангельмана, что свидетельствует о том, что мозг может быть удивительно восприимчив, если утраченный сигнал удастся восстановить.
Короткие генетические направляющие, чтобы прорвать блокировку
Одним из перспективных способов освободить спящий отцовский ген являются антисмысловые олигонуклеотиды, или АСО — короткие синтетические фрагменты генетического материала, которые связываются с конкретными молекулами РНК. При разработке против блокирующей РНК, которая подавляет отцовский UBE3A, эти АСО могут запускать разрушение блокирающей молекулы и позволять отцовской копии снова включиться. Группа сначала протестировала человеческий специфичный АСО в нейронах, полученных из стволовых клеток, выделенных из кожи людей с синдромом Ангельмана и у здоровых добровольцев. Они подтвердили, что по мере созревания в нейроны UBE3A постепенно исчезал в клетках пациентов с Ангельманом, оставаясь выраженным в здоровых клетках. Лечение культур антисмысловым препаратом против блокирующей РНК резко уменьшило уровень блокирающей молекулы, повысило РНК UBE3A и увеличило уровень белка UBE3A до значительной доли нормы — всё это без очевидной токсичности.
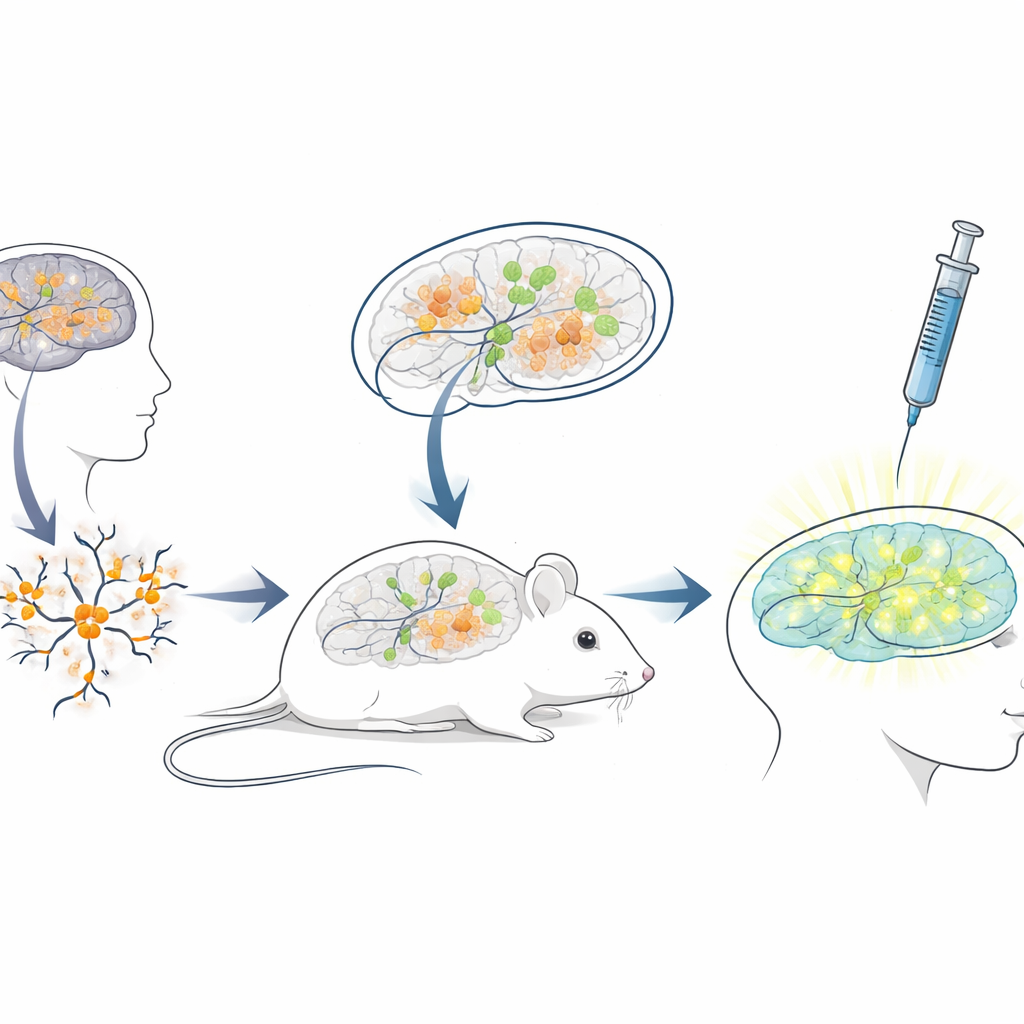
Создание живой мышиной платформы для испытаний человеческих терапий
Поскольку последовательность блокирующей РНК у людей сильно отличается от таковой у мышей, стандартные мышиные модели плохо подходят для тестирования АСО, предназначенных для пациентов. Чтобы преодолеть этот разрыв, исследователи создали платформу «ксенотрансплантации»: они вырастили нейроны из человеческих стволовых клеток с мутациями, вызывающими Ангельмана, а затем пересадили эти молодые человеческие нейроны в мозг новорождённых иммунодефицитных мышей. Трансплантированные клетки оседали в областях мозга, таких как гиппокамп, выживали в течение многих месяцев и развивали ожидаемую потерю UBE3A в ядрах, характерную для Ангельмана. Это создало уникальную ситуацию, когда человеческие больные нейроны живут и созревают внутри мышиного мозга, подвергаясь влиянию естественных мозговых контуров и тем же путям доставки, которые врачи использовали бы у людей.
Испытание препарата, реактивирующего ген, в реалистичных условиях
С этой системой химерного мозг‑мышь‑человек команда имитировала клиническое лечение. Когда мышам было три недели, они ввели антисмысловой препарат в заполненные жидкостью полости мозга, используя тот же общий путь, который применяют для одобренных АСО‑препаратов при других неврологических заболеваниях. Через неделю срезы мозга показали, что пересаженные человеческие нейроны у мышей, получивших препарат, теперь демонстрировали явный ядерный сигнал UBE3A, тогда как у контрольных мышей, получивших только физиологический раствор, сигнала не было. Количественный анализ показал, что почти три четверти человеческих нейронов с мутациями Ангельмана вновь обрели обнаруживаемый UBE3A, а средний сигнал приблизился к уровню здоровых контрольных нейронов. Важно, что мыши хорошо перенесли инъекции — изменений массы тела по сравнению с контролем за период лечения не наблюдалось.
Что это означает для будущих точных лекарств
Эта работа ещё не лечит синдром Ангельмана, но она даёт два ключевых достижения. Во‑первых, она показывает, что человеческий специфичный антисмысловой препарат может надёжно включать молчащий отцовский ген UBE3A в нейронах, полученных от пациентов, как в культурах, так и после интеграции этих нейронов в живой мозг. Во‑вторых, она предоставляет универсальную платформу для тестирования: человеческие нейроны с реальными мутациями пациентов можно вырастить, пересадить в мышей и подвергнуть воздействию кандидатов в лекарства через клинически релевантные маршруты. По мере того как генетическая диагностика редких детских заболеваний мозга становится более частой, такие «персонализированные лаборатории» могут помочь оценивать безопасность и эффективность индивидуальных антисмысловых терапий — даже когда по всему миру лишь несколько пациентов имеют ту или иную мутацию.
Цитирование: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Ключевые слова: Синдром Ангельмана, антисмысловые олигонуклеотиды, ксенотрансплантация, реактивация генов, нейроразвитые расстройства