Clear Sky Science · fr
Un modèle de xénotransplantation pour la réactivation paternelle de UBE3A à l’aide d’oligonucléotides antisens spécifiques de l’humain
Pourquoi réveiller un gène silencieux est important
Le syndrome d’Angelman est une maladie cérébrale rare mais grave qui provoque un retard du développement, des crises d’épilepsie et des troubles sévères de la parole chez l’enfant. Il résulte de la perte d’un gène clé unique, nommé UBE3A, dans les cellules nerveuses. De manière inhabituelle, chaque neurone porte toujours une copie intacte de ce gène héritée du père, mais cette copie est normalement mise en silence. Cette étude explore une façon de « réveiller » cette copie paternelle silencieuse à l’aide d’un médicament génétique ciblé et présente un nouveau modèle animal permettant aux chercheurs de tester en toute sécurité des traitements adaptés à l’humain avant leur application chez les patients.
Une maladie avec un plan de secours caché
Dans la plupart des cellules de l’organisme, le gène UBE3A est actif sur les chromosomes maternel et paternel. Dans les cellules cérébrales, toutefois, seule la copie maternelle est exprimée ; la copie paternelle est bloquée par une longue molécule d’ARN qui se lit dans le sens opposé de l’ADN, comme un train sur une voie de collision empêchant tout autre trafic de passer. Les enfants atteints du syndrome d’Angelman héritent d’une copie maternelle UBE3A défectueuse, ce qui laisse leurs neurones sans protéine UBE3A fonctionnelle malgré la présence d’une copie paternelle intacte. Des travaux antérieurs chez la souris ont montré que rétablir UBE3A tôt dans la vie peut corriger de nombreux symptômes de type Angelman, suggérant que le cerveau peut être étonnamment tolérant si le signal manquant est rétabli.
Utiliser de courts guides génétiques pour couper l’obstacle
Une approche prometteuse pour libérer le gène paternel endormi consiste à utiliser des oligonucléotides antisens, ou ASO : de courts fragments d’acide nucléique synthétiques qui se fixent sur des ARN cibles spécifiques. Conçus contre l’ARN bloqueur qui met en silence UBE3A paternel, ces ASO peuvent déclencher la destruction de cet ARN et permettre à la copie paternelle de se réexprimer. L’équipe a d’abord testé un ASO spécifique de l’humain dans des neurones dérivés de cellules souches issues de prélèvements cutanés de personnes atteintes du syndrome d’Angelman et de volontaires sains. Ils ont confirmé que, à mesure que les cellules mûrissaient en neurones, UBE3A diminuait progressivement dans les cellules Angelman tout en restant élevée dans les cellules saines. Le traitement des cultures par le médicament antisens ciblant l’ARN bloqueur a fortement réduit ce bloqueur, augmenté l’ARN UBE3A et élevé la protéine UBE3A à une fraction substantielle des niveaux normaux — le tout sans toxicité évidente.
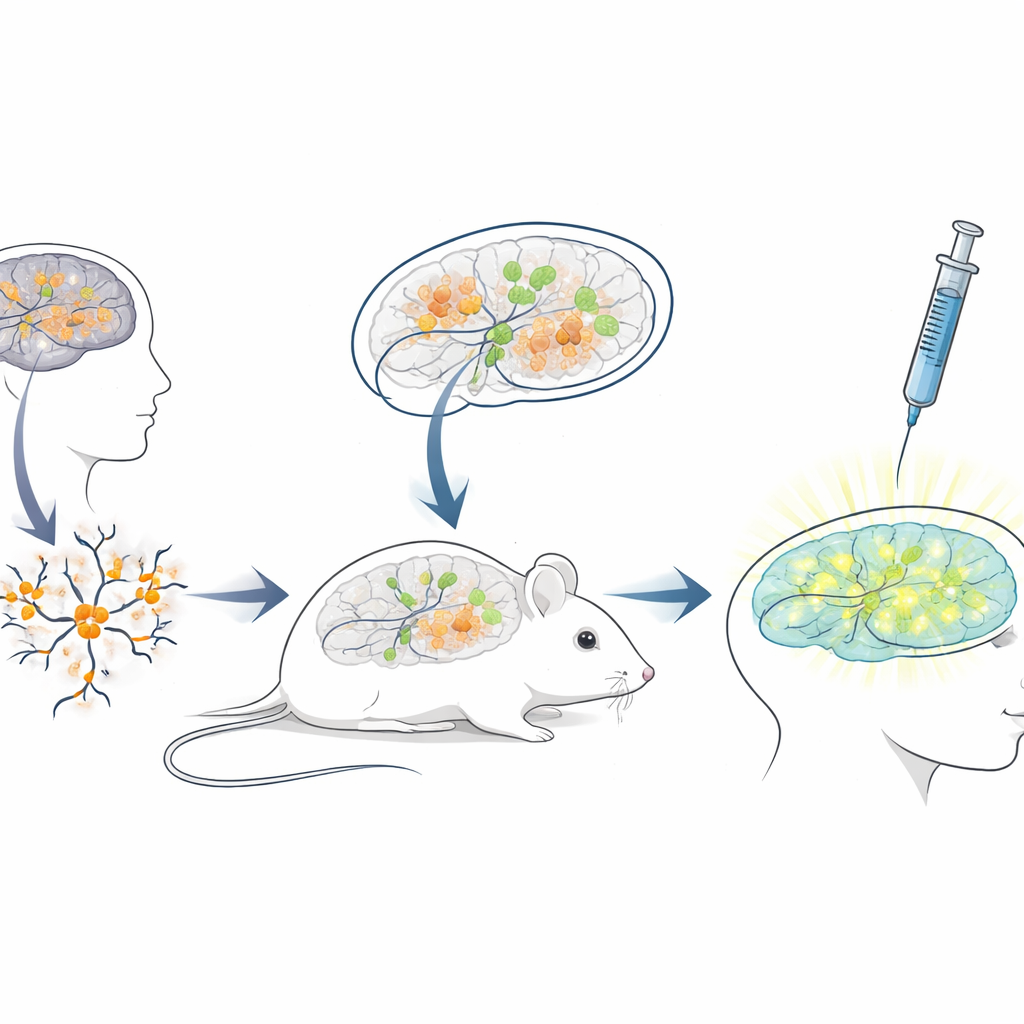
Construire un banc d’essai vivant chez la souris pour des thérapies humaines
Parce que la séquence de l’ARN bloqueur chez l’humain diffère fortement de celle de la souris, les modèles murins standards sont mal adaptés pour tester des ASO destinés aux patients. Pour combler cet écart, les chercheurs ont créé une plate‑forme de « xénotransplantation » : ils ont cultivé des neurones à partir de cellules souches humaines porteuses de mutations d’Angelman, puis ont transplanté ces jeunes neurones humains dans le cerveau de souriceaux nouveau‑nés immunodéficients. Les cellules transplantées se sont intégrées dans des régions cérébrales telles que l’hippocampe, ont survécu plusieurs mois et ont développé la perte attendue de UBE3A dans leurs noyaux, de type Angelman. Cela a créé une situation unique où des neurones humains malades vivent et mûrissent à l’intérieur d’un cerveau de souris, exposés aux circuits neuronaux naturels et aux mêmes voies d’administration que celles utilisées chez l’humain.
Tester le médicament réactivant le gène dans un cadre réaliste
Avec ce système de cerveau chimérique souris–humain en place, l’équipe a reproduit un traitement clinique. Lorsque les souris avaient trois semaines, ils ont injecté le médicament antisens dans les espaces remplis de liquide du cerveau, en utilisant la même voie générale employée pour des ASO approuvés dans d’autres maladies neurologiques. Une semaine plus tard, des coupes cérébrales ont montré que les neurones humains transplantés chez les souris traitées présentaient maintenant un signal nucléaire UBE3A clair, tandis que ceux des souris témoins ayant reçu uniquement une solution saline restaient sombres. L’analyse quantitative a révélé que près des trois quarts des neurones Angelman humains avaient retrouvé une UBE3A détectable, et que le signal moyen approchait celui des neurones témoins sains. Fait important, les souris ont bien toléré les injections, sans différence de poids corporel comparé aux témoins pendant la période de traitement.
Ce que cela signifie pour les médicaments de précision à venir
Ce travail ne guérit pas encore le syndrome d’Angelman, mais il apporte deux avancées cruciales. D’abord, il montre qu’un médicament antisens spécifique de l’humain peut activer de manière fiable le gène paternel UBE3A silencieux dans des neurones dérivés de patients, à la fois en culture et après intégration de ces neurones dans un cerveau vivant. Ensuite, il fournit une plate‑forme de test polyvalente : des neurones humains porteurs de mutations réelles de patients peuvent être cultivés, transplantés dans des souris et exposés à des thérapies candidates via des voies d’administration cliniquement pertinentes. À mesure que les diagnostics génétiques pour les troubles cérébraux infantiles rares se généralisent, de tels « bancs d’essai personnalisés » pourraient aider à évaluer la sécurité et l’efficacité d’ASO sur‑mesure — même lorsque seule une poignée de patients dans le monde partage une mutation donnée.
Citation: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Mots-clés: syndrome d’Angelman, oligonucléotides antisens, xénotransplantation, réactivation génique, troubles du neurodéveloppement