Clear Sky Science · it
Un modello di xenotrapianto per la riattivazione paterna di UBE3A usando oligonucleotidi antisenso specifici per l’uomo
Perché risvegliare un gene silente è importante
La sindrome di Angelman è un raro ma grave disturbo cerebrale che causa ritardo dello sviluppo, crisi epilettiche e gravi problemi del linguaggio nei bambini. Deriva dalla perdita di un singolo gene chiave, chiamato UBE3A, nelle cellule nervose. In modo insolito, ogni neurone conserva ancora una copia sana del gene ereditata dal padre, ma quella copia è normalmente spenta. Questo studio esplora un modo per “risvegliare” quella copia silente con una medicina genetica precisa e introduce un nuovo modello animale che permette ai ricercatori di testare in sicurezza trattamenti su misura per l’uomo prima che arrivino ai pazienti.
Un disturbo con un piano di riserva nascosto
Nella maggior parte delle cellule del corpo, il gene UBE3A è attivo sia sul cromosoma materno sia su quello paterno. Nelle cellule cerebrali, tuttavia, è attiva solo la copia materna; la copia paterna è bloccata da una lunga molecola di RNA che scorre in direzione opposta lungo il DNA, come un treno in rotta di collisione che impedisce ad altro traffico di passare. I bambini con sindrome di Angelman ereditano una UBE3A materna danneggiata, lasciando i loro neuroni privi di proteina UBE3A funzionante nonostante sia presente la copia paterna intatta. Lavori precedenti sui topi hanno mostrato che ripristinare UBE3A precocemente nella vita può correggere molti sintomi simili ad Angelman, suggerendo che il cervello possa essere sorprendentemente indulgente se il segnale mancante viene ripristinato.
Usare brevi guide genetiche per superare il blocco
Un modo promettente per liberare il gene paterno addormentato è tramite oligonucleotidi antisenso, o ASO — brevi frammenti di materiale genetico prodotti in laboratorio che si legano a bersagli RNA specifici. Progettati contro l’RNA bloccante che silenzia UBE3A paterno, questi ASO possono indurre la distruzione di quell’RNA e permettere alla copia paterna di riaccendersi. Il gruppo ha prima testato un ASO specifico per l’uomo in neuroni coltivati da cellule staminali derivate dalla pelle di persone con sindrome di Angelman e di volontari sani. Hanno confermato che, man mano che le cellule maturavano in neuroni, UBE3A scompariva gradualmente nelle cellule Angelman mentre rimaneva elevata in quelle sane. Il trattamento delle colture con il farmaco antisenso contro l’RNA bloccante ha ridotto nettamente il bloccante, aumentato l’RNA di UBE3A e innalzato la proteina UBE3A a una frazione significativa dei livelli normali — il tutto senza tossicità evidente.
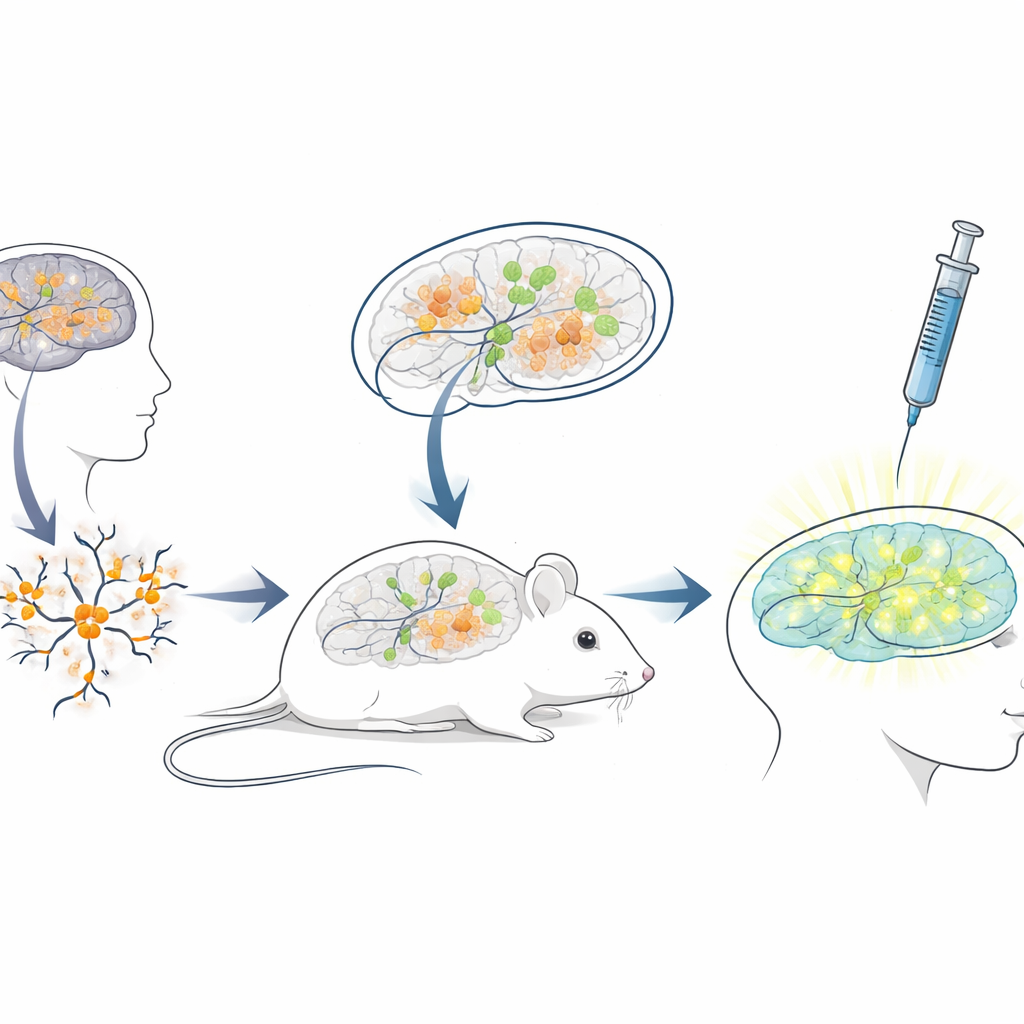
Costruire una piattaforma murina vivente per terapie umane
Poiché la sequenza dell’RNA bloccante negli esseri umani differisce notevolmente da quella nei topi, i modelli murini standard sono poco adatti per testare ASO destinati ai pazienti. Per colmare questa lacuna, i ricercatori hanno creato una piattaforma di “xenotrapianto”: hanno coltivato neuroni da cellule staminali umane portatrici di mutazioni di Angelman, quindi hanno trapiantato questi giovani neuroni umani nei cervelli di topi neonati immunodeficienti. Le cellule trapiantate si sono insediate in regioni cerebrali come l’ippocampo, sono sopravvissute per molti mesi e hanno sviluppato la prevista perdita di UBE3A nei nuclei, tipica di Angelman. Questo ha creato una situazione unica in cui neuroni umani malati vivono e maturano all’interno di un cervello murino, esposti a circuiti cerebrali naturali e alle stesse vie di somministrazione che i medici userebbero nelle persone.
Testare il farmaco riattivante in un contesto realistico
Con questo sistema encefalo chimerico topo‑uomo in funzione, il gruppo ha simulato un trattamento clinico. Quando i topi avevano tre settimane, è stato iniettato il farmaco antisenso negli spazi pieni di fluido del cervello, usando la stessa via generale impiegata per ASO approvati in altre malattie neurologiche. Una settimana dopo, fette cerebrali hanno mostrato che i neuroni umani trapiantati nei topi trattati con il farmaco presentavano ora un chiaro segnale nucleare di UBE3A, mentre quelli nei topi di controllo ricevuti solo soluzione salina restavano scuri. L’analisi quantitativa ha rivelato che quasi tre quarti dei neuroni umani Angelman hanno riacquistato UBE3A rilevabile e il segnale medio si è avvicinato a quello dei neuroni di controllo sani. È importante che i topi abbiano tollerato bene le iniezioni, senza differenze nel peso corporeo rispetto ai controlli nel periodo di trattamento.
Che cosa significa per le future medicine di precisione
Questo lavoro non cura ancora la sindrome di Angelman, ma fornisce due importanti progressi. Primo, dimostra che un farmaco antisenso specifico per l’uomo può riattivare in modo affidabile il gene paterno UBE3A silente in neuroni derivati da pazienti, sia in coltura sia dopo che quei neuroni sono stati integati in un cervello vivente. Secondo, offre una piattaforma di test versatile: neuroni umani portatori di vere mutazioni di pazienti possono essere coltivati, trapiantati in topi ed esposti a terapie candidate tramite vie clinicamente rilevanti. Man mano che le diagnosi genetiche per i rari disturbi cerebrali dell’infanzia diventano più comuni, tali “banchi di prova personalizzati” potrebbero aiutare a valutare la sicurezza e l’efficacia di trattamenti antisenso su misura — anche quando solo una manciata di pazienti nel mondo condivide una determinata mutazione.
Citazione: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Parole chiave: Sindrome di Angelman, oligonucleotidi antisenso, xenotrapianto, riattivazione genica, disturbi del neurosviluppo