Clear Sky Science · en
A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides
Why waking up a silent gene matters
Angelman syndrome is a rare but serious brain disorder that causes developmental delay, seizures, and severe speech problems in children. It stems from the loss of a single key gene, called UBE3A, in nerve cells. Unusually, every neuron still carries a healthy backup copy of this gene from the father, but that copy is normally switched off. This study explores a way to “wake up” that silent backup using a precise genetic medicine, and introduces a new animal model that lets researchers safely test such human‑tailored treatments before they reach patients.
A disorder with a hidden backup plan
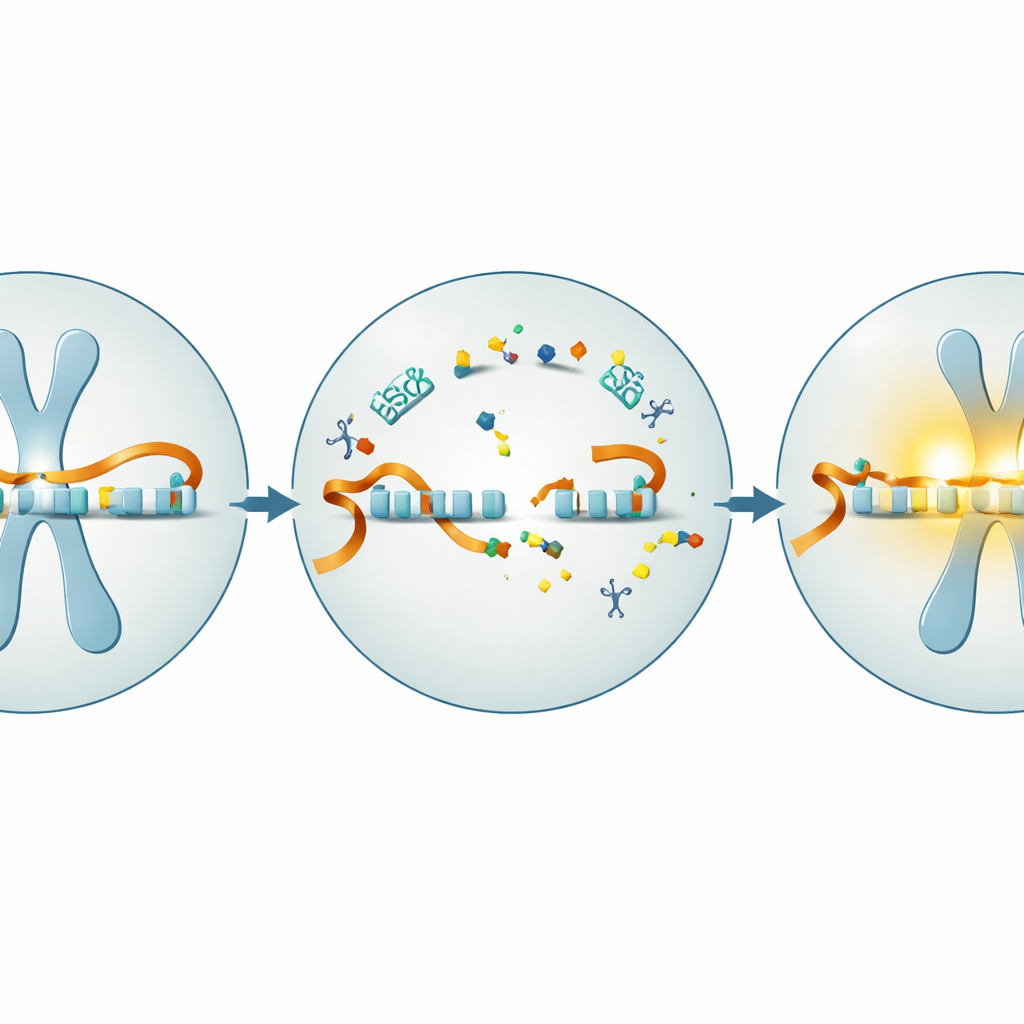
In most of the body’s cells, the UBE3A gene is active on both the mother’s and father’s chromosomes. In brain cells, however, only the maternal copy is turned on; the paternal copy is blocked by a long RNA molecule that runs in the opposite direction along the DNA, like a train on a collision course that prevents other traffic from passing. Children with Angelman syndrome inherit a damaged maternal UBE3A, leaving their neurons without working UBE3A protein even though the intact paternal copy is present. Earlier work in mice showed that restoring UBE3A early in life can rescue many Angelman‑like symptoms, suggesting that the brain may be surprisingly forgiving if the missing signal can be restored.
Using short genetic guides to cut through the blockage
One promising way to free the sleeping paternal gene is through antisense oligonucleotides, or ASOs—short, lab‑made snippets of genetic material that stick to specific RNA targets. When designed against the blocking RNA that silences paternal UBE3A, these ASOs can trigger that RNA’s destruction and allow the paternal gene to turn back on. The team first tested a human‑specific ASO in nerve cells grown from skin‑derived stem cells of people with Angelman syndrome and from healthy volunteers. They confirmed that, as the cells matured into neurons, UBE3A gradually disappeared in Angelman cells while remaining strong in healthy ones. Treating the cultures with the antisense drug against the blocking RNA sharply reduced the blocker, boosted UBE3A RNA, and raised UBE3A protein to a substantial fraction of normal levels—all without obvious toxicity.
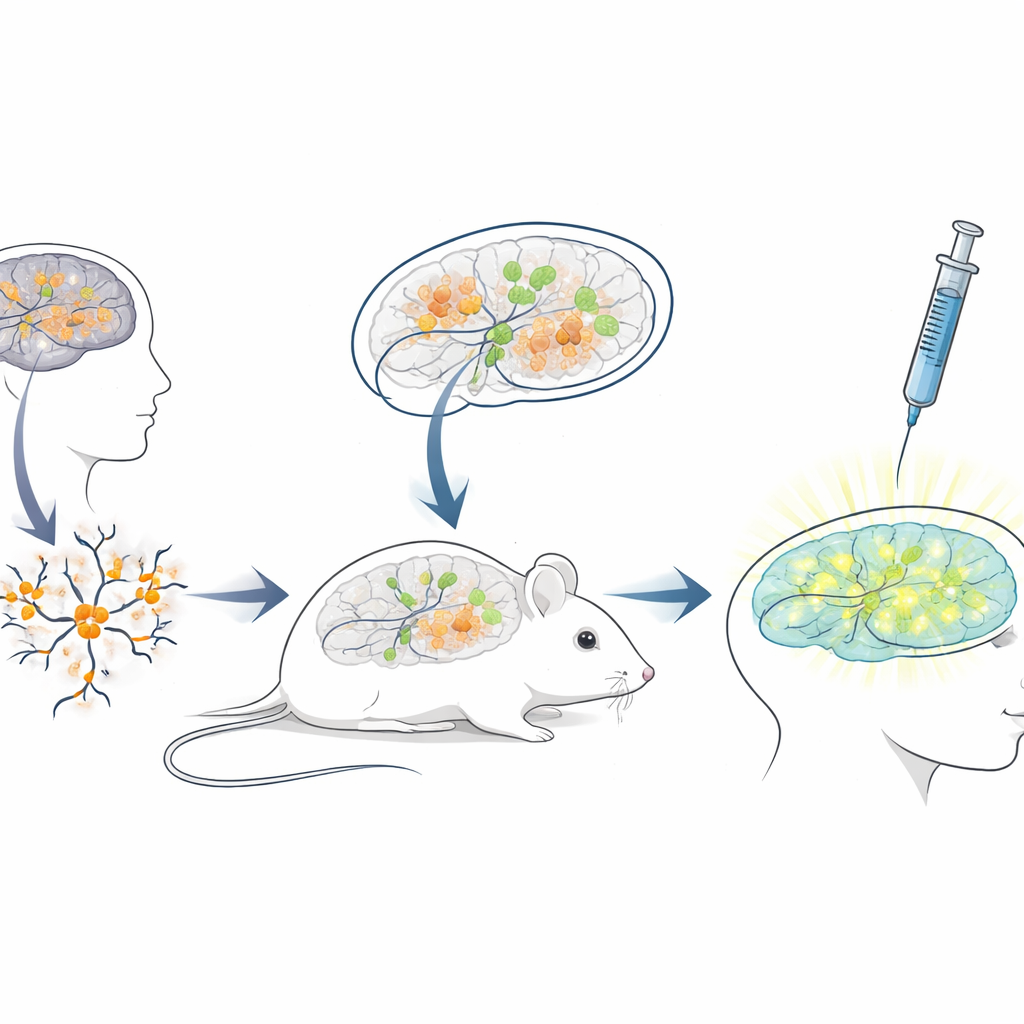
Building a living mouse testbed for human therapies
Because the blocking RNA sequence in humans differs greatly from that in mice, standard mouse models are poorly suited for testing ASOs meant for patients. To bridge this gap, the researchers created a “xenotransplantation” platform: they grew neurons from human stem cells carrying Angelman mutations, then transplanted these young human neurons into the brains of newborn immune‑deficient mice. The transplanted cells settled into brain regions such as the hippocampus, survived for many months, and developed the expected Angelman‑like loss of UBE3A in their nuclei. This set up a unique situation in which human disease neurons live and mature inside a mouse brain, exposed to natural brain circuits and the same delivery routes doctors would use in people.
Testing the gene‑reactivating drug in a realistic setting
With this chimeric mouse–human brain system in place, the team mimicked a clinical treatment. When the mice were three weeks old, they injected the antisense drug into the fluid‑filled spaces of the brain, using the same general route used for approved ASO drugs in other neurological diseases. A week later, brain slices showed that transplanted human neurons in drug‑treated mice now displayed clear nuclear UBE3A signal, while those in control mice receiving only salt solution remained dark. Quantitative analysis revealed that nearly three‑quarters of the human Angelman neurons regained detectable UBE3A, and the average signal approached that of healthy control neurons. Importantly, the mice tolerated the injections well, with no differences in body weight compared to controls over the treatment period.
What this means for future precision medicines
This work does not yet cure Angelman syndrome, but it provides two crucial advances. First, it shows that a human‑specific antisense drug can reliably switch on the silent paternal UBE3A gene in patient‑derived neurons, both in the dish and after those neurons are integrated into a living brain. Second, it delivers a versatile testing platform: human neurons carrying real patient mutations can be grown, transplanted into mice, and exposed to candidate therapies via clinically relevant routes. As genetic diagnoses for rare childhood brain disorders become more common, such “personalized testbeds” may help evaluate safety and effectiveness of tailor‑made antisense treatments—even when only a handful of patients worldwide share a given mutation.
Citation: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Keywords: Angelman syndrome, antisense oligonucleotides, xenotransplantation, gene reactivation, neurodevelopmental disorders