Clear Sky Science · nl
Een xenotransplantatiemodel voor heractivering van vaderlijke UBE3A met mensspecifieke antisense-oligonucleotiden
Waarom het wakker maken van een stilgelegd gen belangrijk is
Het Angelman-syndroom is een zeldzame maar ernstige hersenaandoening die bij kinderen ontwikkelingsachterstand, epileptische toevallen en ernstige spraakproblemen veroorzaakt. De aandoening ontstaat door het verlies van één cruciaal gen, UBE3A, in zenuwcellen. Opmerkelijk genoeg heeft elke neuron nog een gezonde reservekopie van dit gen afkomstig van de vader, maar die kopie is normaal gesproken uitgeschakeld. Deze studie onderzoekt een manier om die stille reservekopie weer te “wekken” met een nauwkeurig genetisch middel, en introduceert een nieuw diermodel waarmee onderzoekers veilig mensgerichte behandelingen kunnen testen voordat ze bij patiënten worden toegepast.
Een aandoening met een verborgen reservestrategie
In de meeste cellen van het lichaam is het UBE3A-gen actief op zowel het chromosoom van de moeder als dat van de vader. In hersencellen daarentegen is alleen de maternale kopie aan; de vaderlijke kopie wordt geblokkeerd door een lang RNA-molecuul dat in de tegengestelde richting over het DNA loopt, als een trein op een botsingskoers die ander verkeer tegenhoudt. Kinderen met het Angelman-syndroom erven een beschadigde maternale UBE3A, waardoor hun neuronen geen werkend UBE3A-eiwit produceren, hoewel de intacte vaderlijke kopie aanwezig is. Eerder werk in muizen toonde aan dat het herstellen van UBE3A vroeg in het leven veel Angelman-achtige symptomen kan verhelpen, wat suggereert dat de hersenen verrassend vergiffenis kunnen tonen als het ontbrekende signaal kan worden hersteld.
Korte genetische gidsen gebruiken om de blokkade door te snijden
Een veelbelovende manier om het slaperige vaderlijke gen vrij te maken is het gebruik van antisense-oligonucleotiden (ASO’s) — korte, kunstmatig gemaakte stukjes genetisch materiaal die zich binden aan specifieke RNA-doelen. Wanneer ze ontworpen zijn tegen het blokkerende RNA dat vaderlijke UBE3A stillegt, kunnen deze ASO’s dat RNA vernietigen en de vaderlijke kopie weer toelaten zich te activeren. Het team testte eerst een mensspecifieke ASO in zenuwcellen die waren gekweekt uit huidafgeleide stamcellen van mensen met Angelman-syndroom en van gezonde vrijwilligers. Ze bevestigden dat, naarmate de cellen uitrijpten tot neuronen, UBE3A geleidelijk verdwenen was in Angelman-cellen terwijl het sterk bleef in gezonde cellen. Behandeling van de kweek met het antisense-middel tegen het blokkerende RNA verminderde de blocker sterk, verhoogde UBE3A-RNA en bracht UBE3A-eiwit naar een substantieel deel van het normale niveau — en dat alles zonder duidelijke toxiciteit.
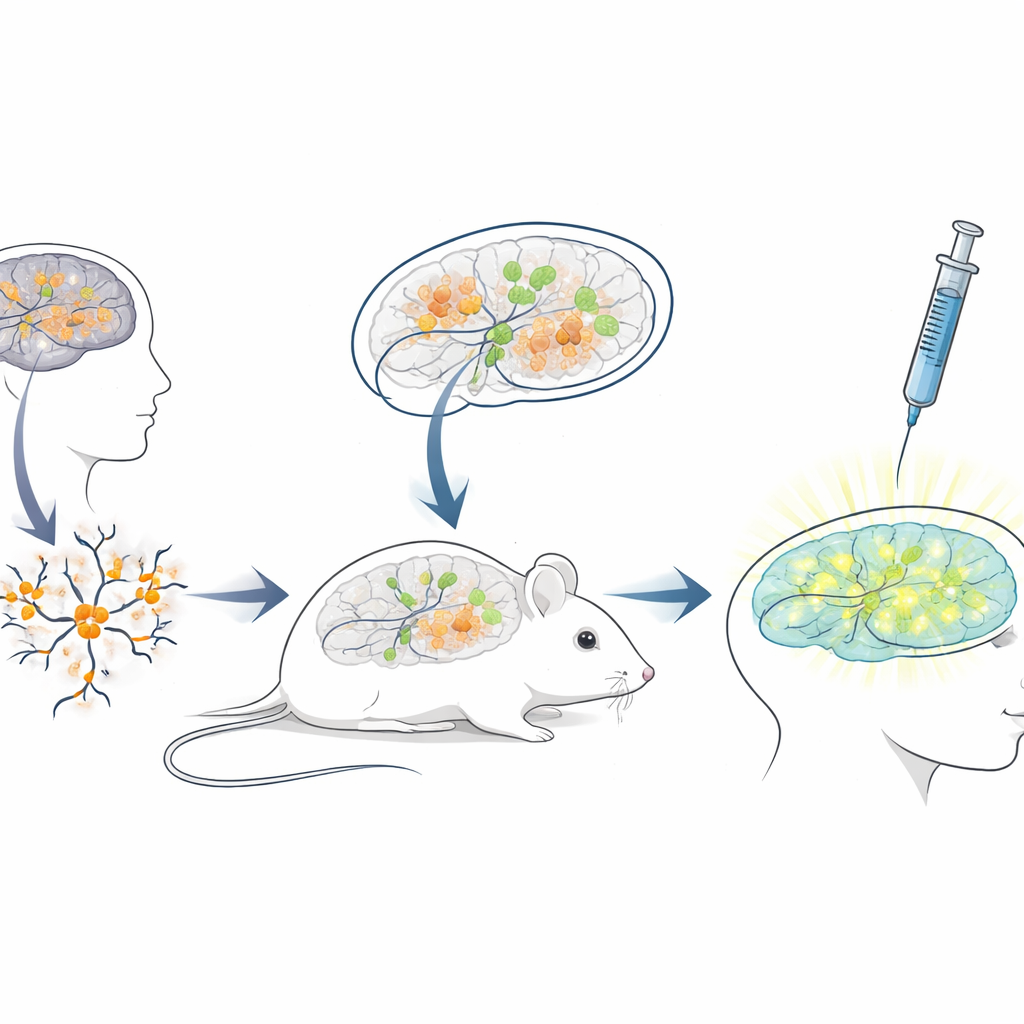
Een levend muistestbed bouwen voor menselijke therapieën
Aangezien de sequentie van het blokkerende RNA bij mensen sterk afwijkt van die bij muizen, zijn standaard muismodellen slecht geschikt voor het testen van ASO’s bedoeld voor patiënten. Om deze kloof te overbruggen creëerden de onderzoekers een “xenotransplantatie”-platform: ze kweekten neuronen uit menselijke stamcellen met Angelman-mutaties en transplanteerden deze jonge menselijke neuronen vervolgens in de hersenen van pasgeboren, immuundeficiënte muizen. De getransplanteerde cellen vestigden zich in hersengebieden zoals de hippocampus, overleefden vele maanden en ontwikkelden het verwachte Angelman-achtige verlies van UBE3A in hun kernen. Dit creëerde een unieke situatie waarin menselijke ziekte-neuronen binnen een muizenbrein leven en rijpen, blootgesteld aan natuurlijke hersencircuits en dezelfde toedieningsroutes die artsen bij mensen zouden gebruiken.
Het gen-heractiverende middel testen in een realistische omgeving
Met dit chimerische muis–mens-hersensysteem op zijn plaats, imiteerde het team een klinische behandeling. Toen de muizen drie weken oud waren, injecteerden ze het antisense-middel in de met vloeistof gevulde ruimten van de hersenen, gebruikmakend van dezelfde algemene route die wordt gebruikt voor goedgekeurde ASO-geneesmiddelen bij andere neurologische aandoeningen. Een week later lieten hersensneden zien dat getransplanteerde menselijke neuronen in geneesmiddel-behandelde muizen nu een duidelijke kernachtige UBE3A-signaal vertoonden, terwijl die in controlemuizen die alleen zoutoplossing kregen donker bleven. Kwantitatieve analyse toonde aan dat bijna driekwart van de menselijke Angelman-neuronen weer detecteerbare UBE3A had gekregen, en het gemiddelde signaal naderde dat van gezonde controleneuronen. Belangrijk is dat de muizen de injecties goed verdroegen, zonder verschillen in lichaamsgewicht vergeleken met controles gedurende de behandelperiode.
Wat dit betekent voor toekomstige precisiegeneesmiddelen
Dit werk geneest het Angelman-syndroom nog niet, maar het levert twee cruciale vooruitgangen. Ten eerste toont het aan dat een mensspecifiek antisense-middel betrouwbaar het stille vaderlijke UBE3A-gen kan inschakelen in patiënt-afgeleide neuronen, zowel in kweek als nadat die neuronen zijn geïntegreerd in een levend brein. Ten tweede biedt het een veelzijdig testplatform: menselijke neuronen met echte patiëntmutaties kunnen worden gekweekt, in muizen worden getransplanteerd en via klinisch relevante routes aan kandidaat-therapieën worden blootgesteld. Naarmate genetische diagnoses voor zeldzame hersenziekten bij kinderen vaker voorkomen, kunnen dergelijke “gepersonaliseerde testbeds” helpen de veiligheid en werkzaamheid van op maat gemaakte antisense-behandelingen te beoordelen — zelfs wanneer wereldwijd slechts een handvol patiënten een bepaalde mutatie deelt.
Bronvermelding: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Trefwoorden: Angelman-syndroom, antisense-oligonucleotiden, xenotransplantatie, genheractivering, neuro-ontwikkelingsstoornissen