Clear Sky Science · de
Ein Xenotransplantationsmodell zur Reaktivierung des paternalen UBE3A mittels menschenspezifischer Antisense‑Oligonukleotide
Warum es wichtig ist, ein stilles Gen zu wecken
Das Angelman‑Syndrom ist eine seltene, aber schwere Hirnerkrankung, die bei Kindern Entwicklungsverzögerung, Anfälle und ausgeprägte Sprechstörungen verursacht. Ursache ist der Verlust eines einzelnen Schlüsselsgens namens UBE3A in Nervenzellen. Ungewöhnlicherweise besitzt jede Neuron weiterhin eine intakte Ersatzkopie dieses Gens vom Vater, doch diese Kopie ist normalerweise abgeschaltet. Diese Studie untersucht eine Methode, diese stille Ersatzkopie mit einer präzisen genetischen Therapie „aufzuwecken“, und stellt ein neues Tiermodell vor, das Forscherinnen und Forschern erlaubt, solche auf Menschen zugeschnittenen Behandlungen sicher zu testen, bevor sie an Patientinnen und Patienten angewendet werden.
Eine Erkrankung mit einem verborgenen Ersatzplan
In den meisten Körperzellen ist das UBE3A‑Gen auf beiden Chromosomen, mütterlich wie väterlich, aktiv. In Gehirnzellen dagegen ist nur die maternale Kopie eingeschaltet; die paternale Kopie wird durch ein langes RNA‑Molekül blockiert, das in entgegengesetzter Richtung entlang der DNA verläuft – wie ein Zug auf Kollisionskurs, der anderen Verkehr am Vorbeifahren hindert. Kinder mit Angelman‑Syndrom erben eine defekte mütterliche UBE3A‑Kopie, sodass ihren Neuronen trotz der intakten väterlichen Kopie funktionierendes UBE3A‑Protein fehlt. Frühere Arbeiten an Mäusen zeigten, dass eine Wiederherstellung von UBE3A früh im Leben viele Angelman‑ähnliche Symptome lindern kann, was darauf hindeutet, dass das Gehirn erstaunlich nachsichtig sein könnte, wenn das fehlende Signal wiederhergestellt wird.
Kurze genetische Führer, die die Blockade durchtrennen
Eine vielversprechende Möglichkeit, das schlafende paternale Gen zu befreien, sind Antisense‑Oligonukleotide (ASOs) – kurze, im Labor gefertigte Abschnitte genetischen Materials, die an spezifische RNA‑Zielsequenzen binden. Werden ASOs gegen die blockierende RNA entworfen, die das paternale UBE3A stummschaltet, können sie den Abbau dieser Blocker‑RNA auslösen und so die Aktivierung des väterlichen Gens ermöglichen. Das Team testete zunächst ein menschenspezifisches ASO in Nervenzellen, die aus hautabgeleiteten Stammzellen von Menschen mit Angelman‑Syndrom und von gesunden Freiwilligen gezüchtet worden waren. Sie bestätigten, dass UBE3A mit der Reifung zu Neuronen in den Angelman‑Zellen allmählich verschwand, während es in gesunden Zellen erhalten blieb. Die Behandlung der Kulturen mit dem antisense Therapeutikum gegen die blockierende RNA reduzierte diesen Blocker deutlich, erhöhte die UBE3A‑RNA und hob das UBE3A‑Protein auf einen beträchtlichen Bruchteil des Normalwerts – und das ohne offensichtliche Toxizität.
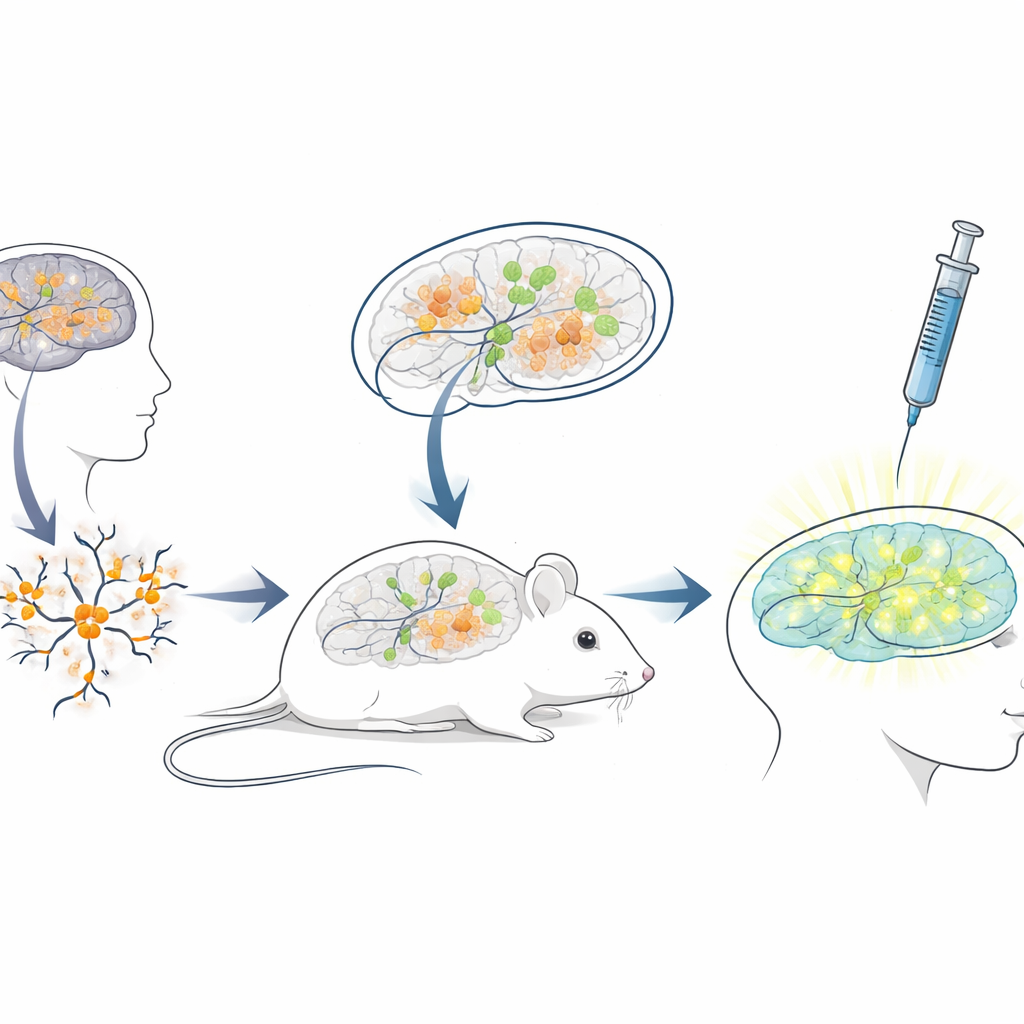
Ein lebender Maus‑Testkörper für menschliche Therapien
Da die Sequenz der blockierenden RNA beim Menschen sich stark von der der Maus unterscheidet, eignen sich Standardmausmodelle nur begrenzt zum Testen von ASOs, die für Patienten gedacht sind. Um diese Lücke zu schließen, entwickelten die Forschenden eine „Xenotransplantations“‑Plattform: Sie züchteten Neuronen aus menschlichen Stammzellen mit Angelman‑Mutationen und transplantierten diese jungen humanen Neuronen in die Gehirne neugeborener, immundefizienter Mäuse. Die transplantierten Zellen siedelten sich in Hirnregionen wie dem Hippocampus an, überlebten über viele Monate und zeigten den erwarteten Angelman‑ähnlichen Verlust von UBE3A in ihren Zellkernen. So entstand eine einzigartige Situation, in der menschliche Krankheitsneurone innerhalb eines Mausgehirns leben und reifen, exponiert gegenüber natürlichen Schaltkreisen des Gehirns und denselben Verabreichungswegen, die Ärztinnen und Ärzte auch beim Menschen nutzen würden.
Prüfung des genreaktivierenden Medikaments in einer realistischen Umgebung
Mit diesem chimären Maus‑Mensch‑Gehirnsystem führte das Team eine klinisch realistische Behandlungssimulation durch. Als die Mäuse drei Wochen alt waren, injizierten sie das antisense Medikament in die flüssigkeitsgefüllten Räume des Gehirns, entlang derselben allgemeinen Route, die für zugelassene ASO‑Medikamente bei anderen neurologischen Erkrankungen verwendet wird. Eine Woche später zeigten Hirnschnitte, dass transplantierte menschliche Neurone in medikamentenbehandelten Mäusen nun ein deutliches nukleäres UBE3A‑Signal aufwiesen, während diejenigen in Kontrollmäusen, die nur Kochsalzlösung erhielten, dunkel blieben. Die quantitative Analyse ergab, dass nahezu drei Viertel der humanen Angelman‑Neurone wieder nachweisbares UBE3A zurückgewannen und das durchschnittliche Signal sich dem von gesunden Kontrollneuronen annäherte. Wichtig war außerdem, dass die Mäuse die Injektionen gut tolerierten und es während des Behandlungszeitraums keine Unterschiede im Körpergewicht gegenüber den Kontrollen gab.
Was das für künftige Präzisionsmedikamente bedeutet
Diese Arbeit heilt das Angelman‑Syndrom noch nicht, liefert aber zwei entscheidende Fortschritte. Erstens zeigt sie, dass ein menschenspezifisches Antisense‑Medikament zuverlässig das stille paternale UBE3A‑Gen in patientenabgeleiteten Neuronen aktivieren kann – sowohl in der Zellkultur als auch nachdem diese Neurone in ein lebendes Gehirn integriert wurden. Zweitens stellt sie eine vielseitige Prüfplattform bereit: Menschliche Neurone mit echten Patientenmutationen können gezüchtet, in Mäuse transplantiert und über klinisch relevante Wege Kandidatentherapien ausgesetzt werden. Da genetische Diagnosen seltener kindlicher Hirnerkrankungen zunehmend häufiger gestellt werden, könnten solche „personalisierten Testfelder“ helfen, Sicherheit und Wirksamkeit maßgeschneiderter Antisense‑Behandlungen zu bewerten – selbst wenn weltweit nur eine Handvoll Patientinnen und Patienten dieselbe Mutation teilen.
Zitation: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Schlüsselwörter: Angelman‑Syndrom, Antisense‑Oligonukleotide, Xenotransplantation, Genreaktivierung, neuroentwicklungsstörungen