Clear Sky Science · pt
Um modelo de xenotransplante para reativação do UBE3A paterno usando oligonucleotídeos antissenso específicos para humanos
Por que despertar um gene silenciado importa
A síndrome de Angelman é um transtorno cerebral raro, porém grave, que causa atraso no desenvolvimento, convulsões e sérios problemas de fala em crianças. Ela decorre da perda de um único gene essencial, chamado UBE3A, nas células nervosas. De maneira incomum, cada neurônio carrega uma cópia de reserva saudável desse gene herdada do pai, mas essa cópia costuma estar desligada. Este estudo explora uma forma de “acordar” essa cópia silenciosa usando uma terapia genética precisa, e apresenta um novo modelo animal que permite aos pesquisadores testar com segurança tratamentos feitos sob medida para humanos antes de chegarem aos pacientes.
Um transtorno com um plano alternativo oculto
Na maior parte das células do corpo, o gene UBE3A está ativo tanto nos cromossomos maternos quanto paternos. Nas células do cérebro, porém, apenas a cópia materna é expressa; a cópia paterna fica bloqueada por uma longa molécula de RNA que corre na direção oposta ao longo do DNA, como um trem em rota de colisão que impede a passagem de outro tráfego. Crianças com síndrome de Angelman herdam um UBE3A materno danificado, deixando seus neurônios sem proteína UBE3A funcional, mesmo com a cópia paterna intacta presente. Trabalhos anteriores em camundongos mostraram que restaurar o UBE3A cedo na vida pode resgatar muitos sintomas semelhantes aos da Angelman, sugerindo que o cérebro pode ser surpreendentemente permissivo se o sinal ausente puder ser restabelecido.
Usando guias genéticos curtos para cortar o bloqueio
Uma abordagem promissora para liberar o gene paterno adormecido é o uso de oligonucleotídeos antissenso, ou ASOs — pequenos trechos de material genético sintético que se ligam a RNAs alvos específicos. Quando projetados contra o RNA bloqueador que silencia o UBE3A paterno, esses ASOs podem induzir a degradação desse RNA e permitir que o gene paterno volte a ser expresso. A equipe testou primeiro um ASO específico para humanos em células nervosas obtidas de células‑tronco derivadas da pele de pessoas com síndrome de Angelman e de voluntários saudáveis. Confirmaram que, conforme as células amadureciam em neurônios, o UBE3A desaparecia gradualmente nas células Angelman enquanto permanecia presente nas saudáveis. O tratamento das culturas com o medicamento antissenso contra o RNA bloqueador reduziu fortemente o bloqueador, aumentou o RNA de UBE3A e elevou a proteína UBE3A a uma fração substancial dos níveis normais — tudo isso sem toxicidade óbvia.
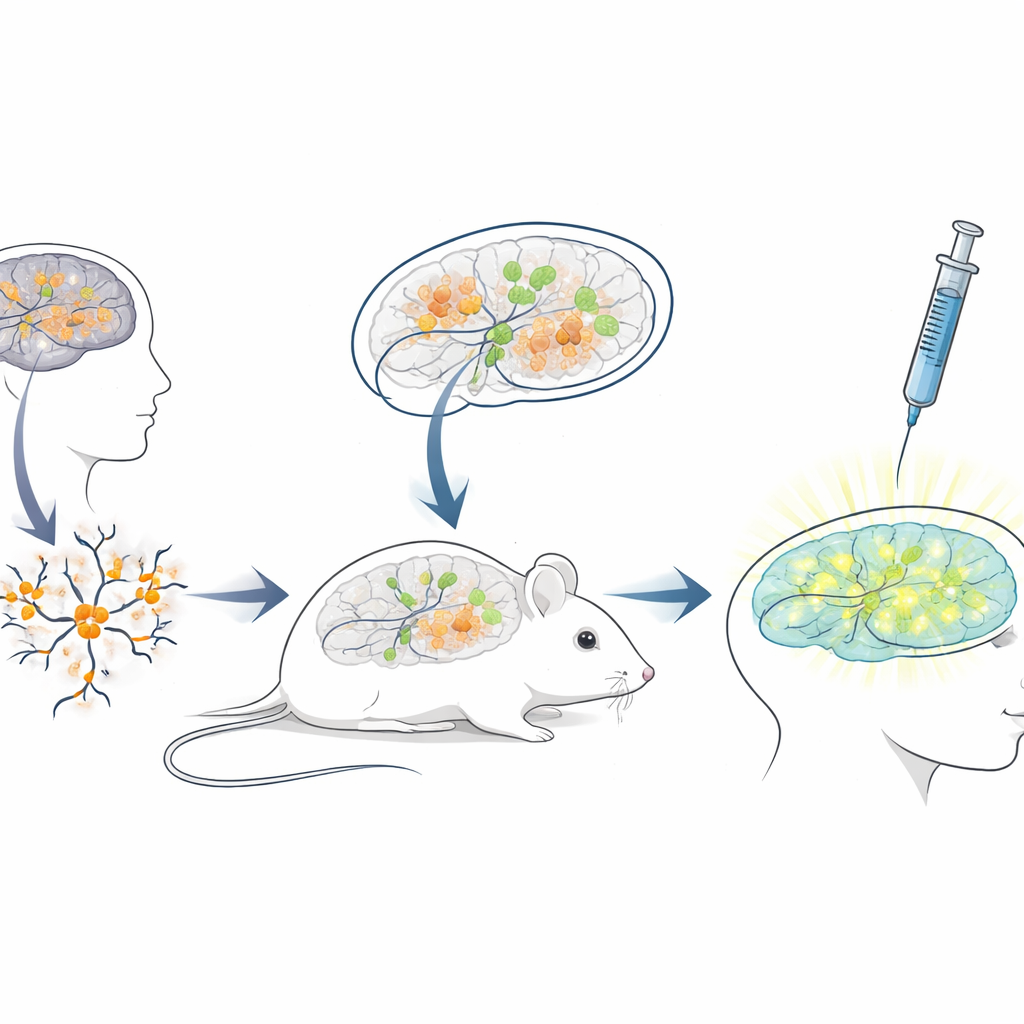
Construindo um banco de testes vivo em camundongos para terapias humanas
Como a sequência do RNA bloqueador em humanos difere muito da dos camundongos, modelos murinos padrão são pouco adequados para testar ASOs destinados a pacientes. Para preencher essa lacuna, os pesquisadores criaram uma plataforma de “xenotransplante”: cultivaram neurônios a partir de células‑tronco humanas com mutações de Angelman e então transplantaram esses neurônios jovens no cérebro de camundongos recém‑nascidos com deficiência imunológica. As células transplantadas se alojaram em regiões cerebrais como o hipocampo, sobreviveram por muitos meses e desenvolveram a esperada perda de UBE3A no núcleo, característica da Angelman. Isso criou uma situação única em que neurônios humanos doentes vivem e amadurecem dentro de um cérebro de camundongo, expostos a circuitos cerebrais naturais e às mesmas rotas de administração que os clínicos usariam em pessoas.
Testando a droga reativadora do gene em um cenário realista
Com esse sistema quimérico camundongo–humano estabelecido, a equipe simulou um tratamento clínico. Quando os camundongos tinham três semanas de idade, injetaram o medicamento antissenso nos espaços preenchidos por líquido do cérebro, usando a mesma via geral empregada para ASOs aprovados em outras doenças neurológicas. Uma semana depois, cortes cerebrais mostraram que os neurônios humanos transplantados em camundongos tratados com o medicamento exibiam agora sinal nuclear claro de UBE3A, enquanto aqueles em camundongos controle que receberam apenas solução salina permaneciam escuros. Análises quantitativas revelaram que quase três quartos dos neurônios humanos Angelman recuperaram UBE3A detectável, e o sinal médio se aproximou do observado em neurônios de controle saudáveis. Importante, os camundongos toleraram bem as injeções, sem diferenças no peso corporal em comparação com os controles durante o período de tratamento.
O que isso significa para futuras medicinas de precisão
Este trabalho ainda não cura a síndrome de Angelman, mas fornece dois avanços cruciais. Primeiro, demonstra que um medicamento antissenso específico para humanos pode ligar de forma confiável o gene UBE3A paterno silencioso em neurônios derivados de pacientes, tanto em cultura quanto depois que esses neurônios são integrados a um cérebro vivo. Segundo, entrega uma plataforma versátil de testes: neurônios humanos com mutações reais de pacientes podem ser cultivados, transplantados em camundongos e expostos a terapias candidatas por vias clinicamente relevantes. À medida que diagnósticos genéticos para raros transtornos cerebrais infantis se tornam mais comuns, esses “bancos de testes personalizados” podem ajudar a avaliar a segurança e a eficácia de tratamentos antissenso sob medida — mesmo quando apenas um punhado de pacientes no mundo compartilha uma dada mutação.
Citação: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Palavras-chave: Síndrome de Angelman, oligonucleotídeos antissenso, xenotransplante, reativação gênica, transtornos do neurodesenvolvimento