Clear Sky Science · pl
Model ksenotransplantacji do reaktywacji ojcowskiego UBE3A przy użyciu przeciwstawnych oligonukleotydów specyficznych dla człowieka
Dlaczego obudzenie wyciszonego genu ma znaczenie
Zespół Angelmana to rzadkie, ale poważne zaburzenie mózgu, które powoduje opóźnienie rozwoju, napady padaczkowe i ciężkie problemy z mową u dzieci. Wynika z utraty jednego kluczowego genu o nazwie UBE3A w komórkach nerwowych. Co nietypowe, każdy neuron wciąż ma zdrową zapasową kopię tego genu pochodzącą od ojca, lecz ta kopia jest zwykle wyłączona. Badanie to analizuje sposób „obudzenia” tej uśpionej kopii przy użyciu precyzyjnego leku genetycznego i wprowadza nowy model zwierzęcy, który pozwala badaczom bezpiecznie testować takie zabiegi dopasowane do człowieka, zanim trafią do pacjentów.
Zaburzenie z ukrytym planem zapasowym
W większości komórek organizmu gen UBE3A jest aktywny zarówno na chromosomie matczynym, jak i ojcowskim. W komórkach mózgu jednak tylko matczyna kopia jest włączona; ojcowska kopia jest zablokowana przez długą cząsteczkę RNA, która przebiega w przeciwnym kierunku względem DNA — jak pociąg na kursie kolizyjnym, który uniemożliwia ruch innym. Dzieci z zespołem Angelmana dziedziczą uszkodzoną matczyną kopię UBE3A, co pozostawia ich neurony bez działającego białka UBE3A, mimo obecności nieuszkodzonej kopii ojcowskiej. Wcześniejsze badania na myszach wykazały, że przywrócenie UBE3A we wczesnym okresie życia może uratować wiele objawów podobnych do Angelmana, co sugeruje, że mózg może być zaskakująco podatny na naprawę, jeśli brakujący sygnał zostanie przywrócony.
Użycie krótkich przewodników genetycznych, by przerwać zator
Jednym z obiecujących sposobów uwolnienia uśpionego genu ojcowskiego są przeciwstawne oligonukleotydy (ASO) — krótkie, laboratoryjnie syntetyzowane fragmenty materiału genetycznego, które wiążą się z określonymi cząsteczkami RNA. Skierowane przeciwko blokującemu RNA, który ucisza ojcowskie UBE3A, ASO mogą wywołać degradację tego blokera i pozwolić na ponowne włączenie genu ojcowskiego. Zespół najpierw przetestował ASO specyficzne dla człowieka w neuronach wyhodowanych ze skórnych komórek macierzystych osób z zespołem Angelmana oraz od zdrowych ochotników. Potwierdzili, że w miarę dojrzewania komórek w neurony, UBE3A stopniowo zanika w komórkach Angelmana, podczas gdy w komórkach zdrowych pozostaje silny. Leczenie kultur przeciwstawnym lekiem skierowanym przeciw blokującemu RNA znacząco zmniejszyło ilość blokera, zwiększyło RNA UBE3A i podniosło poziom białka UBE3A do znaczącej części poziomu normalnego — wszystko to bez wyraźnej toksyczności.
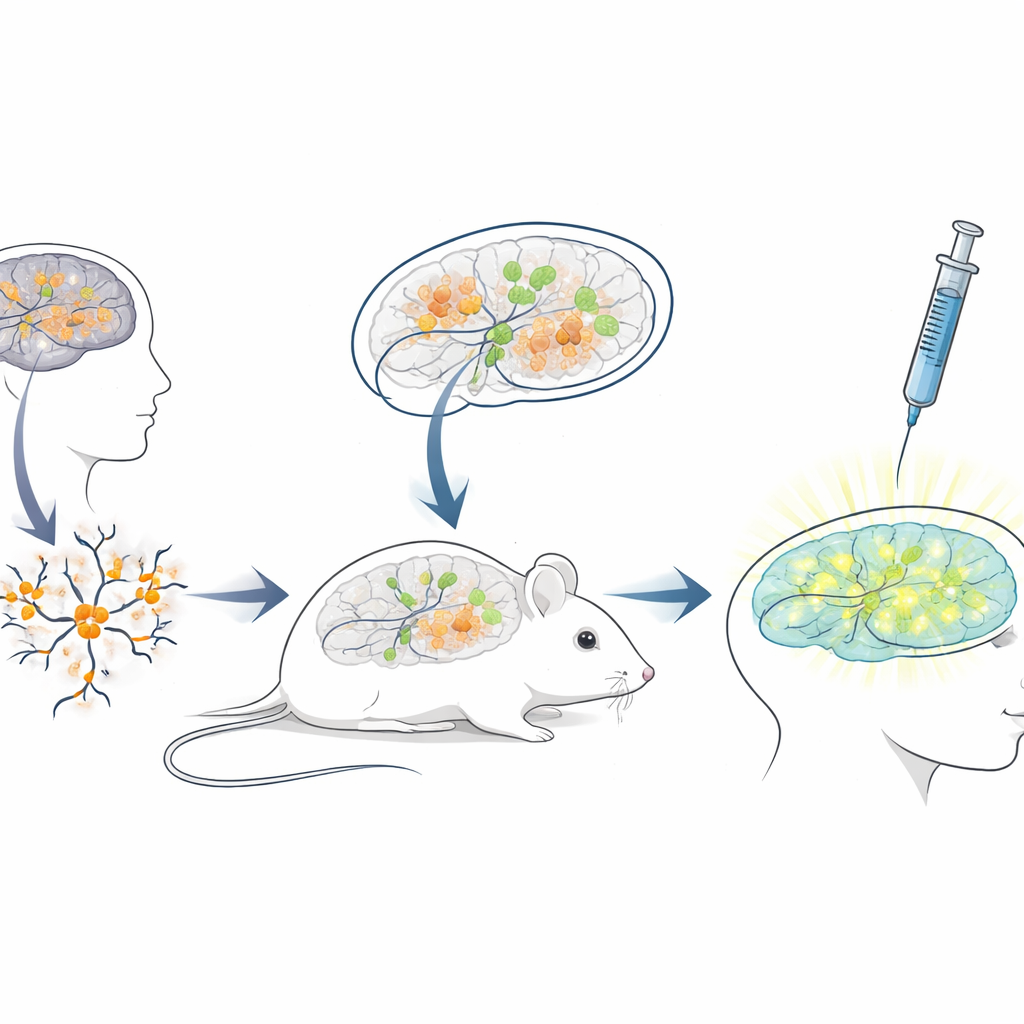
Budowanie żywego modelu myszy do testów terapii ludzkich
Ponieważ sekwencja blokującego RNA u ludzi różni się znacznie od tej u myszy, standardowe modele mysie słabo nadają się do testowania ASO przeznaczonych dla pacjentów. Aby zniwelować tę różnicę, naukowcy stworzyli platformę „ksenotransplantacyjną”: hodowali neurony z ludzkich komórek macierzystych noszących mutacje Angelmana, a następnie przeszczepiali te młode ludzkie neurony do mózgów nowo narodzonych myszy o upośledzonej odporności. Przeszczepione komórki osiedlały się w obszarach mózgu, takich jak hipokamp, przetrwały wiele miesięcy i rozwinęły oczekiwane, przypominające Angelmana, wyciszenie UBE3A w jądrach. Utworzyło to unikalną sytuację, w której ludzkie neurony chorobowe żyją i dojrzewają wewnątrz mózgu myszy, wystawione na działanie naturalnych obwodów mózgowych i tych samych dróg podawania, jakich lekarze używaliby u ludzi.
Testowanie leku reaktywującego gen w realistycznym środowisku
Z tym chimerycznym systemem myszo‑ludzkiego mózgu zespół naśladował leczenie kliniczne. Gdy myszy miały trzy tygodnie, wstrzyknęli lek przeciwstawny do płynem wypełnionych przestrzeni mózgu, używając tej samej ogólnej drogi podania co zatwierdzone leki ASO w innych chorobach neurologicznych. Tydzień później wycinki mózgu pokazały, że przeszczepione ludzkie neurony u myszy leczonych lekiem wykazywały wyraźny sygnał jądrowego UBE3A, podczas gdy te u myszy kontrolnych otrzymujących jedynie roztwór soli pozostały ciemne. Analiza ilościowa wykazała, że niemal trzy czwarte ludzkich neuronów Angelmana odzyskało wykrywalne UBE3A, a średni sygnał zbliżył się do poziomu neuronów kontrolnych zdrowych. Co ważne, myszy dobrze tolerowały wstrzyknięcia, bez różnic w masie ciała w porównaniu do kontroli w okresie leczenia.
Co to oznacza dla przyszłych leków precyzyjnych
Ta praca jeszcze nie leczy zespołu Angelmana, ale dostarcza dwóch kluczowych postępów. Po pierwsze, pokazuje, że przeciwstawny lek specyficzny dla człowieka może niezawodnie włączyć uśpiony ojcowski gen UBE3A w neuronach pochodzących od pacjentów, zarówno in vitro, jak i po zintegrowaniu tych neuronów z żywym mózgiem. Po drugie, dostarcza wszechstronnej platformy testowej: ludzkie neurony niosące rzeczywiste mutacje pacjentów mogą być hodowane, przeszczepiane do myszy i eksponowane na kandydackie terapie drogami odpowiednimi klinicznie. W miarę jak diagnostyka genetyczna rzadkich dziecięcych zaburzeń mózgu staje się powszechniejsza, takie „spersonalizowane poligony testowe” mogą pomóc ocenić bezpieczeństwo i skuteczność szytych na miarę terapii przeciwstawnych — nawet gdy na świecie tylko garstka pacjentów ma daną mutację.
Cytowanie: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Słowa kluczowe: Zespół Angelmana, przeciwstawne oligonukleotydy, ksenotransplantacja, reaktywacja genów, zaburzenia neurorozwojowe