Clear Sky Science · sv
En xenotransplantationsmodell för reaktivering av paternell UBE3A med hjälp av människaspecifika antisense-oligonukleotider
Varför det betyder något att väcka en tyst gen
Angelmans syndrom är en sällsynt men allvarlig hjärnsjukdom som orsakar utvecklingsförsening, anfall och svåra talproblem hos barn. Den beror på förlusten av en enda viktig gen, kallad UBE3A, i nervcellerna. Ovanligt nog har varje neuron fortfarande en intakt reservkopia av denna gen från fadern, men den kopian är normalt avstängd. Denna studie undersöker ett sätt att “väcka” den tysta reservkopian med ett precisionsgenetiskt läkemedel och presenterar en ny djurmodell som gör det möjligt för forskare att säkert testa sådana människaanpassade behandlingar innan de når patienter.
En sjukdom med en dold reservplan
I de flesta kroppens celler är UBE3A-genen aktiv på både moderns och faderns kromosomer. I hjärnceller däremot är endast den maternella kopian påslagen; den paternella kopian blockeras av ett långt RNA som löper i motsatt riktning längs DNA, som ett tåg på kollisionskurs som hindrar annan trafik från att passera. Barn med Angelmans syndrom ärver en skadad maternell UBE3A, vilket lämnar deras neuroner utan fungerande UBE3A-protein även om den intakta paternella kopian finns. Tidigare arbete i möss visade att återställande av UBE3A tidigt i livet kan rädda många Angelman-liknande symtom, vilket tyder på att hjärnan kan vara förvånansvärt förlåtande om den saknade signalen kan återställas.
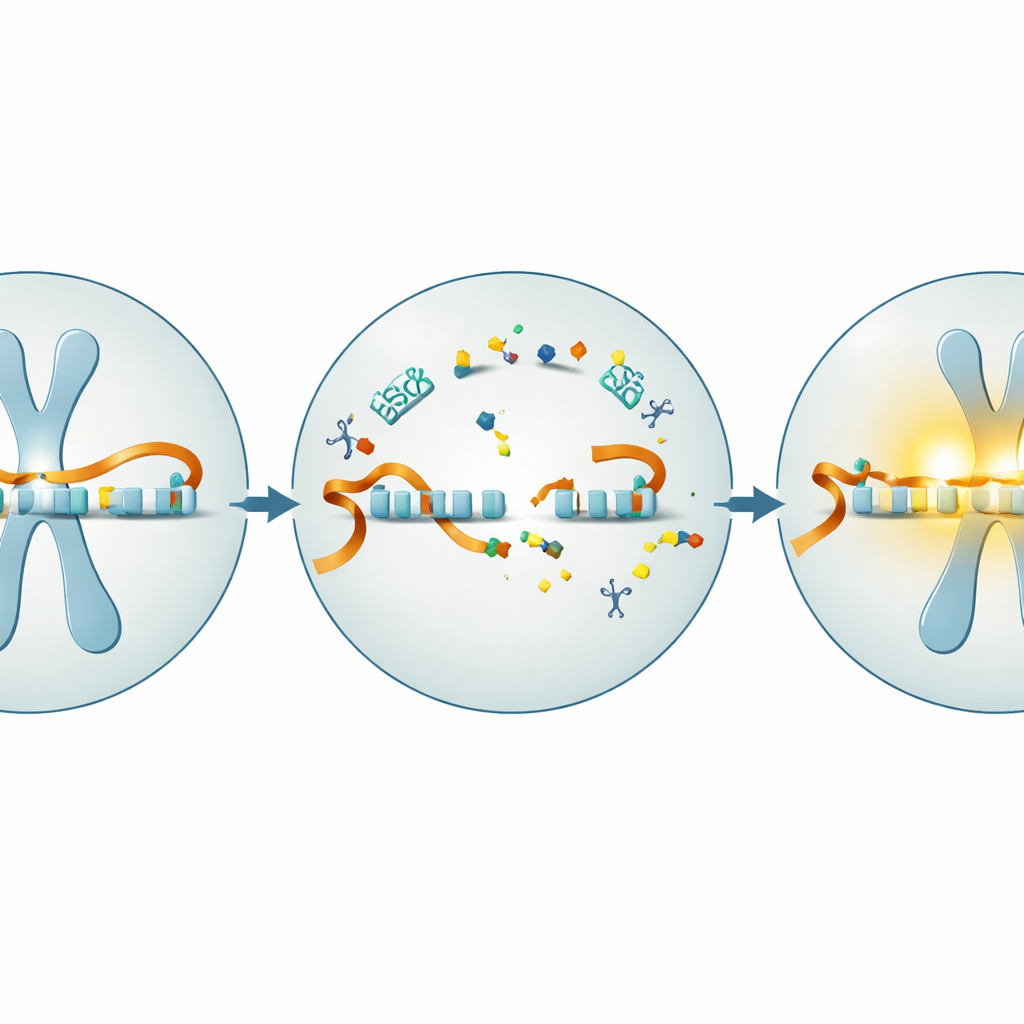
Använda korta genetiska vägvisare för att skära igenom blockeringen
Ett lovande sätt att frigöra den sovande paternella genen är genom antisense-oligonukleotider, eller ASO:er — korta, laboratorietillverkade bitar av genetiskt material som fäster vid specifika RNA-mål. När de är utformade mot det blockerande RNA som tystar paternell UBE3A kan dessa ASO:er utlösa nedbrytning av blockern och tillåta att den paternella genen slås på igen. Teamet testade först en människaspecifik ASO i nervceller som odlats från hudderiverade stamceller från personer med Angelmans syndrom och från friska frivilliga. De bekräftade att när cellerna mognade till neuroner försvann UBE3A gradvis i Angelman-celler medan den förblev stark i friska celler. Behandling av odlingarna med det antisense-läkemedlet mot det blockerande RNA:t minskade blockern markant, ökade UBE3A-RNA och höjde UBE3A-proteinet till en betydande del av normala nivåer — allt utan tydlig toxicitet.
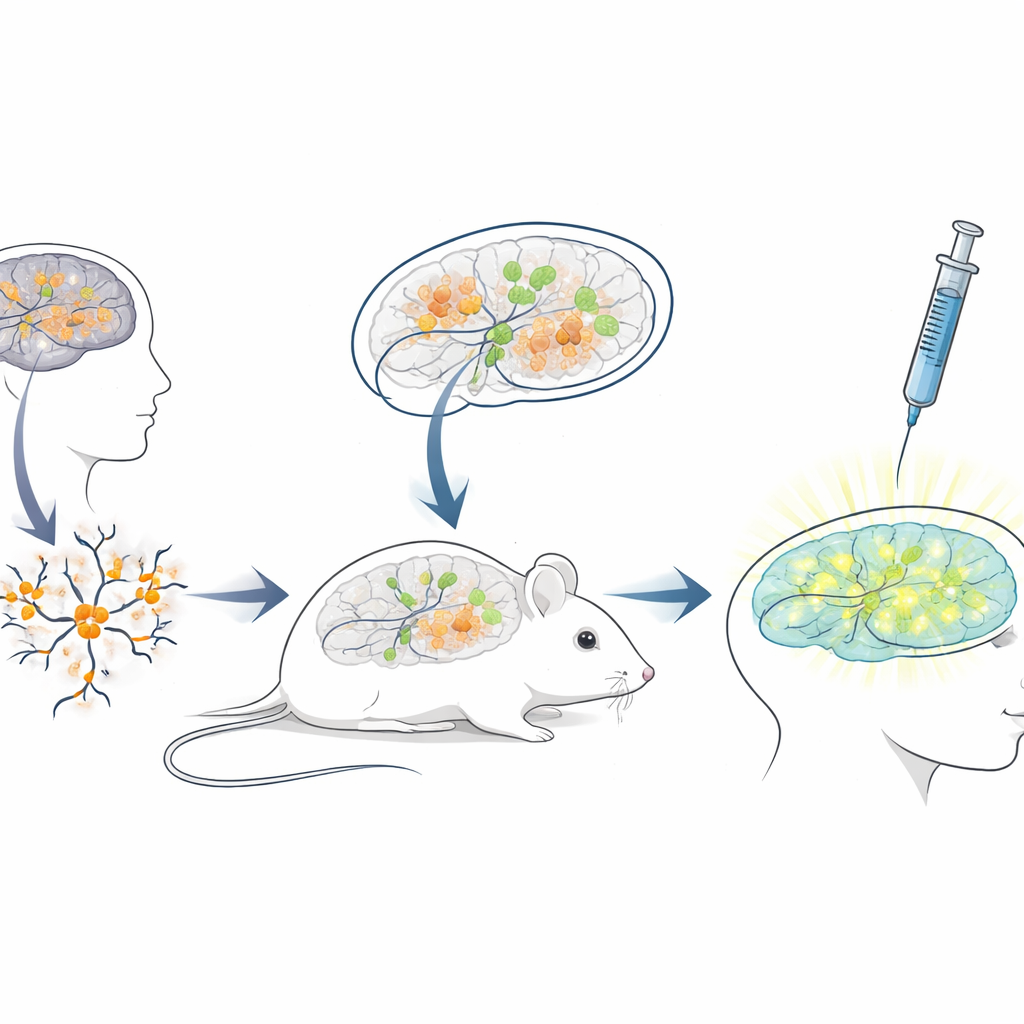
Bygga en levande musplattform för humana terapier
Där det blockerande RNA-sekvensen hos människor skiljer sig mycket från den hos möss är vanliga musmodeller dåligt lämpade för att testa ASO:er avsedda för patienter. För att överbrygga detta gap skapade forskarna en ”xenotransplantations”plattform: de odlade neuroner från humana stamceller som bar Angelman-mutationer och transplanterade sedan dessa unga humana neuroner in i hjärnorna på nyfödda immundefekta möss. De transplanterade cellerna bosatte sig i hjärnregioner som hippocampus, överlevde i många månader och utvecklade den förväntade Angelman-liknande förlusten av UBE3A i sina kärnor. Detta skapade en unik situation där humana sjukdomsneuroner lever och mognar inuti en mushjärna, utsatta för naturliga hjärnkretsar och samma administrationsvägar som läkare skulle använda hos människor.
Testa det genreaktiverande läkemedlet i en realistisk miljö
Med detta kimeriska mus‑humana hjärnsystem på plats efterliknade teamet en klinisk behandling. När mössen var tre veckor gamla injicerade de det antisense-läkemedlet i hjärnans vätskefyllda utrymmen, med samma allmänna rout som används för godkända ASO-läkemedel vid andra neurologiska sjukdomar. En vecka senare visade hjärnsnitt att transplanterade humana neuroner i läkemedelsbehandlade möss nu uppvisade tydlig nukleär UBE3A-signal, medan de i kontrollmöss som endast fick saltslösning förblev mörka. Kvantitativ analys visade att nästan tre fjärdedelar av de humana Angelman-neuronerna återfick detekterbar UBE3A, och den genomsnittliga signalen närmade sig den hos friska kontrollneuroner. Viktigt är att mössen tolererade injektionerna väl, utan skillnad i kroppsvikt jämfört med kontroller under behandlingsperioden.
Vad detta innebär för framtidens precisionsläkemedel
Detta arbete botar ännu inte Angelmans syndrom, men det ger två viktiga framsteg. För det första visar det att ett människaspecifikt antisense-läkemedel pålitligt kan slå på den tysta paternella UBE3A-genen i patientderiverade neuroner, både i odling och efter att dessa neuroner integrerats i en levande hjärna. För det andra levererar det en mångsidig testplattform: humana neuroner med verkliga patientmutationer kan odlas, transplanteras i möss och exponeras för kandidatbehandlingar via kliniskt relevanta vägar. När genetiska diagnoser för sällsynta barnhjärnsjukdomar blir vanligare kan sådana "personaliserade testbäddar" hjälpa till att utvärdera säkerhet och effektivitet hos skräddarsydda antisense‑behandlingar — även när bara en handfull patienter i världen delar en viss mutation.
Citering: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Nyckelord: Angelmans syndrom, antisense-oligonukleotider, xenotransplantation, genreaktivering, neurodevelopmentala störningar