Clear Sky Science · es
Un modelo de xenotrasplante para la reactivación del UBE3A paterno mediante oligonucleótidos antisentido específicos humanos
Por qué importa despertar un gen silencioso
El síndrome de Angelman es un trastorno cerebral raro pero grave que provoca retraso del desarrollo, convulsiones y severos problemas del habla en niños. Se debe a la pérdida de un único gen clave, llamado UBE3A, en las neuronas. De forma inusual, cada neurona conserva una copia sana de este gen heredada del padre, pero esa copia suele estar apagada. Este estudio explora una forma de “despertar” esa copia silenciosa mediante un fármaco genético preciso y presenta un nuevo modelo animal que permite a los investigadores probar de forma segura tratamientos diseñados para humanos antes de que lleguen a los pacientes.
Un trastorno con un plan de respaldo oculto
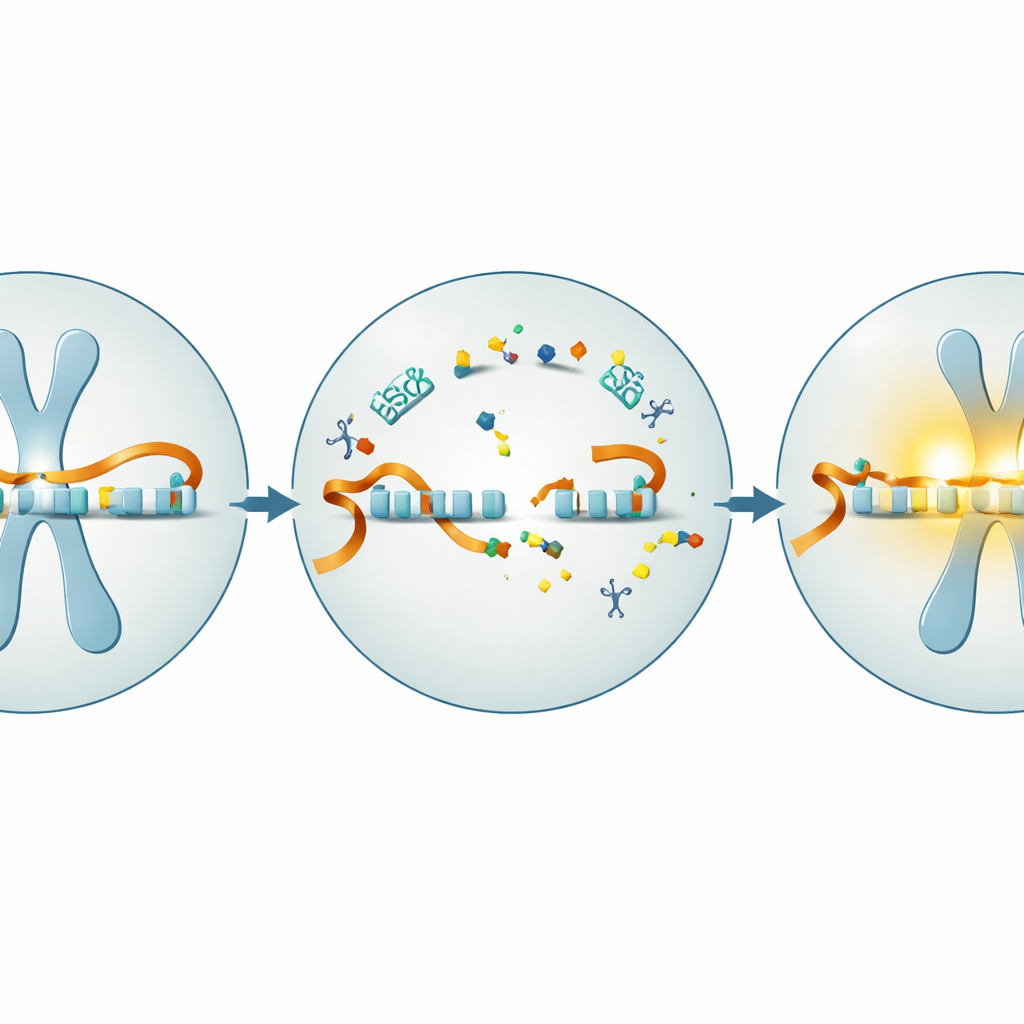
En la mayoría de las células del cuerpo, el gen UBE3A está activo en los cromosomas materno y paterno. En las células cerebrales, sin embargo, sólo se activa la copia materna; la copia paterna está bloqueada por una larga molécula de ARN que se transcribe en la dirección opuesta a lo largo del ADN, como un tren en curso de colisión que impide que otro tráfico pase. Los niños con síndrome de Angelman heredan una UBE3A materna dañada, lo que deja a sus neuronas sin la proteína UBE3A funcional aunque la copia paterna intacta esté presente. Trabajos previos en ratones mostraron que restaurar UBE3A temprano en la vida puede rescatar muchos síntomas similares a los del Angelman, lo que sugiere que el cerebro puede ser sorprendentemente tolerante si se restablece la señal faltante.
Usar guías genéticas cortas para abrir paso al bloqueo
Una vía prometedora para liberar el gen paterno dormido son los oligonucleótidos antisentido, u ASO, fragmentos cortos de material genético sintético que se unen a dianas específicas de ARN. Diseñados contra el ARN bloqueador que silencia UBE3A paterno, estos ASO pueden provocar la destrucción de ese ARN y permitir que el gen paterno vuelva a activarse. El equipo probó primero un ASO específico para humanos en neuronas cultivadas a partir de células madre derivadas de la piel de personas con síndrome de Angelman y de voluntarios sanos. Confirmaron que, a medida que las células maduraban hacia neuronas, UBE3A desaparecía gradualmente en las células de Angelman mientras se mantenía en las sanas. El tratamiento de los cultivos con el fármaco antisentido dirigido contra el ARN bloqueador redujo drásticamente el bloqueador, aumentó el ARN de UBE3A y elevó la proteína UBE3A hasta una fracción sustancial de los niveles normales, todo ello sin toxicidad aparente.
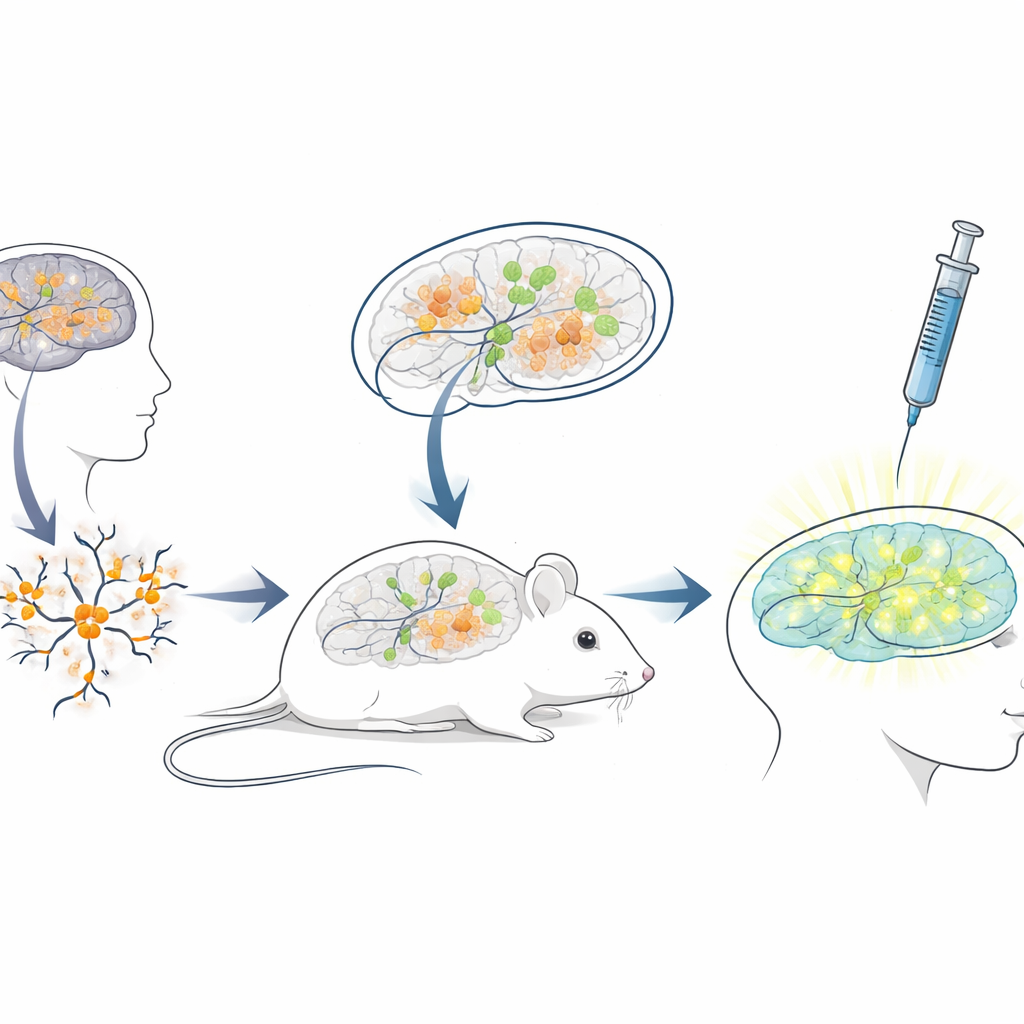
Construir un banco de pruebas vivo en ratones para terapias humanas
Dado que la secuencia del ARN bloqueador en humanos difiere mucho de la de los ratones, los modelos murinos estándar son poco adecuados para probar ASO destinados a pacientes. Para salvar esta brecha, los investigadores crearon una plataforma de “xenotrasplante”: hicieron crecer neuronas a partir de células madre humanas portadoras de mutaciones de Angelman y luego trasplantaron estas jóvenes neuronas humanas en los cerebros de ratones recién nacidos inmunodeficientes. Las células trasplantadas se asentaron en regiones cerebrales como el hipocampo, sobrevivieron durante muchos meses y desarrollaron la esperada pérdida tipo Angelman de UBE3A en sus núcleos. Esto generó una situación única en la que neuronas humanas enfermas viven y maduran dentro de un cerebro de ratón, expuestas a circuitos cerebrales naturales y a las mismas rutas de administración que los médicos usarían en personas.
Probar el fármaco que reactiva el gen en un escenario realista
Con este sistema cerebral quimérico ratón–humano establecido, el equipo simuló un tratamiento clínico. Cuando los ratones tenían tres semanas, inyectaron el fármaco antisentido en los espacios llenos de líquido del cerebro, usando la misma vía general empleada para ASO aprobados en otras enfermedades neurológicas. Una semana después, cortes cerebrales mostraron que las neuronas humanas trasplantadas en ratones tratados con el fármaco exhibían ahora una señal nuclear clara de UBE3A, mientras que las de los ratones control que recibieron sólo solución salina permanecían oscuras. El análisis cuantitativo reveló que casi tres cuartas partes de las neuronas humanas de Angelman recuperaron UBE3A detectable, y la señal media se acercó a la de las neuronas control sanas. Es importante destacar que los ratones toleraron bien las inyecciones, sin diferencias en el peso corporal en comparación con los controles durante el periodo de tratamiento.
Qué significa esto para futuras medicinas de precisión
Este trabajo aún no cura el síndrome de Angelman, pero aporta dos avances cruciales. Primero, demuestra que un fármaco antisentido específico para humanos puede activar de forma fiable el gen UBE3A paterno silencioso en neuronas derivadas de pacientes, tanto en cultivo como después de que esas neuronas se integren en un cerebro vivo. Segundo, proporciona una plataforma de ensayo versátil: neuronas humanas con mutaciones reales de pacientes pueden cultivarse, trasplantarse a ratones y exponerse a terapias candidatas mediante vías clínicamente relevantes. A medida que los diagnósticos genéticos de los trastornos cerebrales infantiles raros se vuelvan más comunes, estos “bancos de pruebas personalizados” podrían ayudar a evaluar la seguridad y la eficacia de tratamientos antisentido a medida, incluso cuando sólo un puñado de pacientes en todo el mundo comparta una mutación determinada.
Cita: Smeenk, H., Lendemeijer, B., Buurma, M.G. et al. A xenotransplantation model for reactivation of paternal UBE3A using human-specific antisense oligonucleotides. Sci Rep 16, 11443 (2026). https://doi.org/10.1038/s41598-026-41197-9
Palabras clave: Síndrome de Angelman, oligonucleótidos antisentido, xenotrasplante, reactivación génica, trastornos del neurodesarrollo