Clear Sky Science · zh
跨模型与任务量化蛋白质表征的不确定性
为什么蛋白质人工智能的可靠性很重要
人工智能已成为观察蛋白质这个无形世界的强大显微镜。现代“蛋白质语言模型”能够仅凭氨基酸序列推测蛋白质的三维形态及其可能的行为。这些模型已在新药设计和理解疾病突变方面发挥作用。但有一个隐含的问题:它们很少告诉我们应当在多大程度上信任它们内部创建的表征。本文通过一个看似简单但影响深远的问题来填补这一空白:当模型把蛋白质转换为一团数字时,我们如何判断这团数字是真实生物学信息的反映,还是仅仅是噪声?
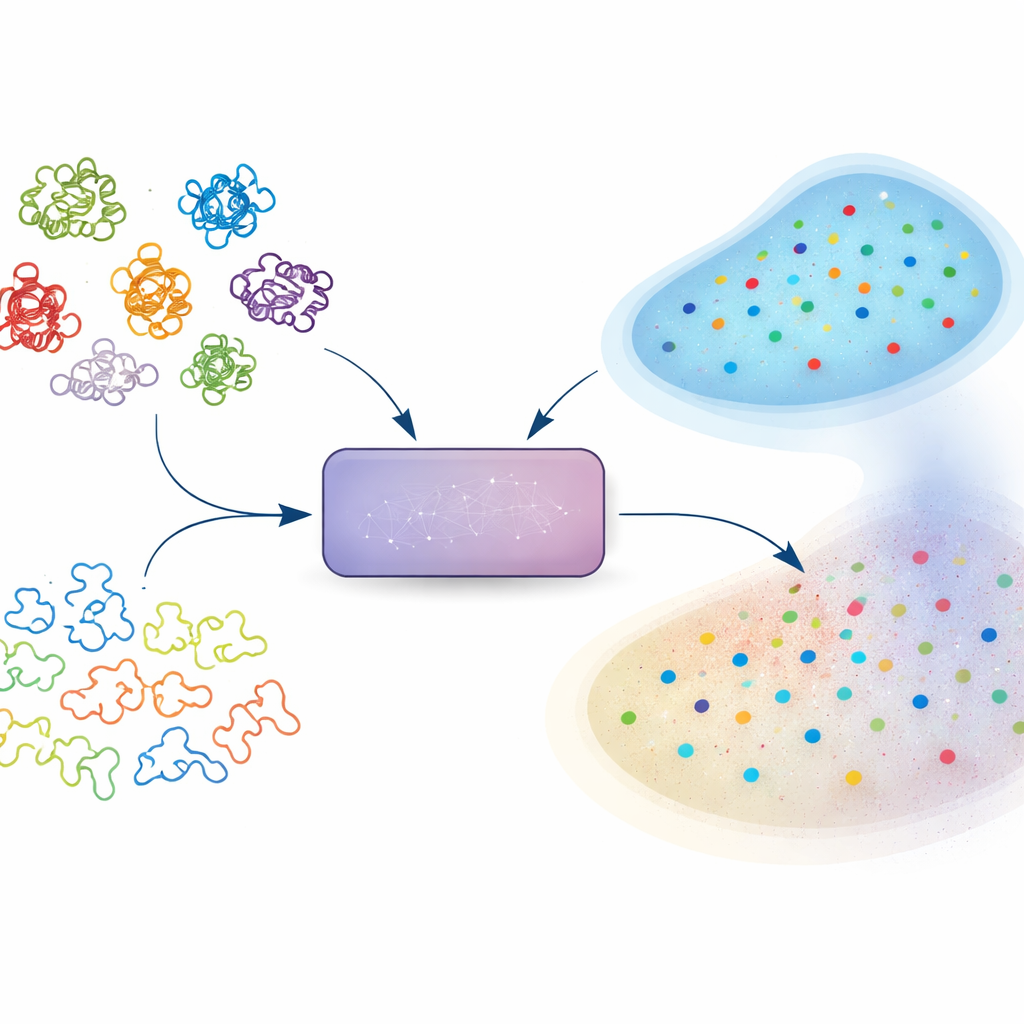
从句子到蛋白质
语言模型最初用于处理人类文本,学习词与词之间的关系并预测句子接下来的内容。同样的思想现在驱动着把蛋白质和 DNA 序列当作长“词”来阅读的模型。对每个蛋白质,模型会产生一个“嵌入”——在高维空间中的一个点,用以总结模型对该蛋白质的认知。这些嵌入被用于许多下游任务,例如预测结构、功能以及突变的影响。然而,与熟悉的预测得分或置信度不同,嵌入通常被直接接受:当模型输出一个向量时,用户往往会信任它,即便在模型训练时几乎未见过的蛋白质区域也是如此。
识别模型何时在猜测
作者提出了一种无需修改底层模型即可估计嵌入可信度的实用方法。他们的核心想法是向模型提供一组故意打乱的蛋白质序列,这些序列保留了基本的成分但失去了所有有意义的生物学模式。这些合成序列充当“垃圾场”——当没有真实信号可学时模型会产生什么的参考。对于任意真实蛋白质,该方法检查在模型内部空间中与之最接近的邻居有多少来自这个垃圾场。如果附近有许多点来自被打乱的序列,则该蛋白质的表征可能学习不足或模糊。作者将这个来自垃圾场邻居的比例称为随机邻居得分(Random Neighbor Score,RNS)。
将不确定性与真实世界性能联系起来
为了检验 RNS 是否反映生物学上的重要信息,研究团队使用包括 ESM-2 和 ProtT5 在内的多种最先进模型,分析了大量蛋白质结构和序列。他们发现,结构预测准确的蛋白质通常具有较低的 RNS——即它们的嵌入远离垃圾场。相反,结构预测不佳的蛋白质位于真实序列与打乱序列重叠的区域。该模式在不同模型和任务中一致存在。当他们观察更应用化的问题,如预测哪些氨基酸残基在三维中发生接触或分配二级结构时,随着 RNS 增加,准确性明显下降。换句话说,嵌入越不确定(RNS 越高),下游预测就越不可靠。
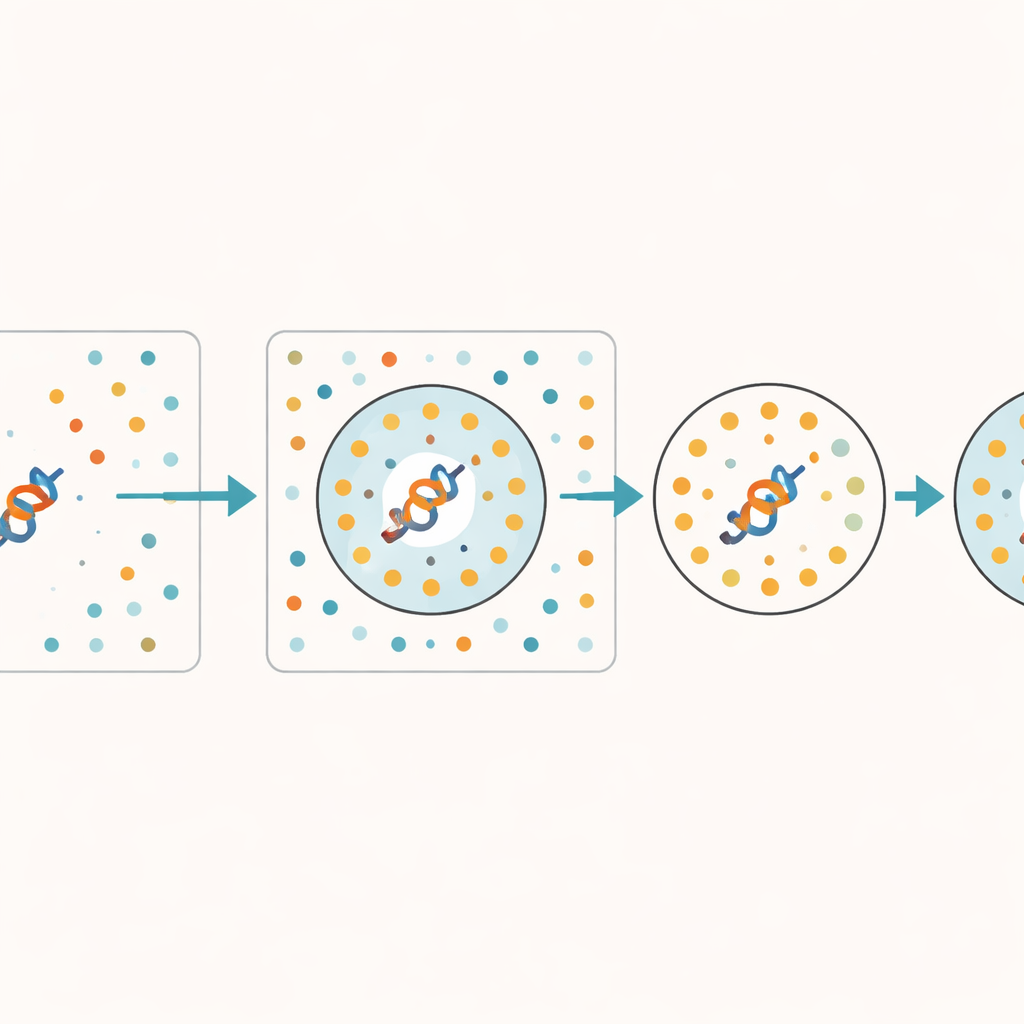
蛋白质空间中的盲点
RNS 还揭示了模型在表征蛋白质宇宙不同部分时的系统性盲点。内在无序区域——缺乏稳定结构的灵活片段——的 RNS 一直高于结构良好的域,表明模型在处理这些滑溜的序列时更吃力。即便在研究较多的人类蛋白质组中,也有相当一部分蛋白质的 RNS 非零,表明它们并未被流行模型很好地捕捉。令人惊讶的是,更大的模型并不总是更好:一个以结构为重点的大模型对许多人类蛋白质可能比一个更小、更加通用的模型更不确定。对于新发现的宏基因组蛋白质,甚至那些为显得逼真而计算设计的“幻觉”蛋白质,低 RNS 表明当模式一致时,模型可以自信地推广到其训练数据之外。
更好的筛选带来更好的生物学洞见
作者接着测试了基于 RNS 的筛查如何影响一个临床相关任务:预测人类蛋白质中单个字母变化是否可能破坏其功能或导致疾病。当他们将分析限制在 RNS 低的蛋白质上——这些蛋白质的嵌入似乎是可靠的——模型性能显著提升,通常能在有害和中性变体之间做出强有力的区分。对于 RNS 高的蛋白质,预测准确性下降到接近抛硬币的水平。这支持了这样一种观点:不可靠的嵌入会悄然限制任何基于它们构建的下游工具所能达到的最佳准确度,而不管采用了多么巧妙的训练策略。
这对在生物学中使用人工智能意味着什么
对非专家而言,结论是并非所有由人工智能得到的蛋白质表征都同样值得信赖,而现在可以对这种可靠性进行量化。随机邻居得分充当对嵌入的简单、与模型无关的健康检查:低分表明某个蛋白质位于其他有生物学意义序列之间,而高分则暗示它向随机相似物的垃圾场漂移。通过在进行结构预测、功能注释或优先排序疾病变体之前基于 RNS 对蛋白质进行筛选或加权,研究者可以把注意力集中在模型真正“理解”数据的区域。正如没有科学家会在不留意的情况下使用模糊的显微镜一样,这项工作主张每个蛋白质语言模型都应配备一种内建的方法来衡量其对生物学内部视图的清晰度。
引用: Prabakaran, R., Bromberg, Y. Quantifying uncertainty in protein representations across models and tasks. Nat Methods 23, 796–804 (2026). https://doi.org/10.1038/s41592-026-03028-7
关键词: 蛋白质语言模型, 嵌入可靠性, 表征不确定性, 变体效应预测, 内在无序蛋白质