Clear Sky Science · fr
Quantifier l’incertitude dans les représentations protéiques à travers modèles et tâches
Pourquoi la fiabilité en IA protéique compte
L’intelligence artificielle est devenue un microscope puissant pour le monde invisible des protéines. Les « modèles de langage protéique » modernes peuvent deviner l’apparence 3D d’une protéine et son comportement probable rien qu’à partir de sa séquence de briques constitutives. Ces modèles aident déjà à concevoir de nouveaux médicaments et à comprendre les mutations liées aux maladies. Mais un problème caché subsiste : ils indiquent rarement combien il faut se fier aux représentations internes qu’ils produisent. Cet article comble cette lacune en posant une question simple aux conséquences importantes : quand un modèle transforme une protéine en un nuage de nombres, comment savoir si ce nuage reflète réellement la biologie ou s’il n’est que du bruit ?
Des phrases aux protéines
Les modèles de langage ont d’abord été développés pour traiter le texte humain, en apprenant comment les mots se relient et en prédisant la suite d’une phrase. Les mêmes idées alimentent désormais des modèles qui lisent les séquences de protéines et d’ADN comme s’il s’agissait de longs mots. Pour chaque protéine, le modèle produit un « embedding » — un point dans un espace de haute dimension censé résumer ce que le modèle sait de cette protéine. Ces embeddings sont ensuite utilisés pour de nombreuses tâches en aval, comme prédire la structure, la fonction ou l’impact des mutations. Pourtant, contrairement aux scores de prédiction ou mesures de confiance familiers, les embeddings sont généralement pris pour acquis : si le modèle renvoie un vecteur, les utilisateurs ont tendance à lui faire confiance, même dans des régions de l’espace protéique que le modèle a peu vues pendant l’entraînement.
Repérer quand le modèle devine
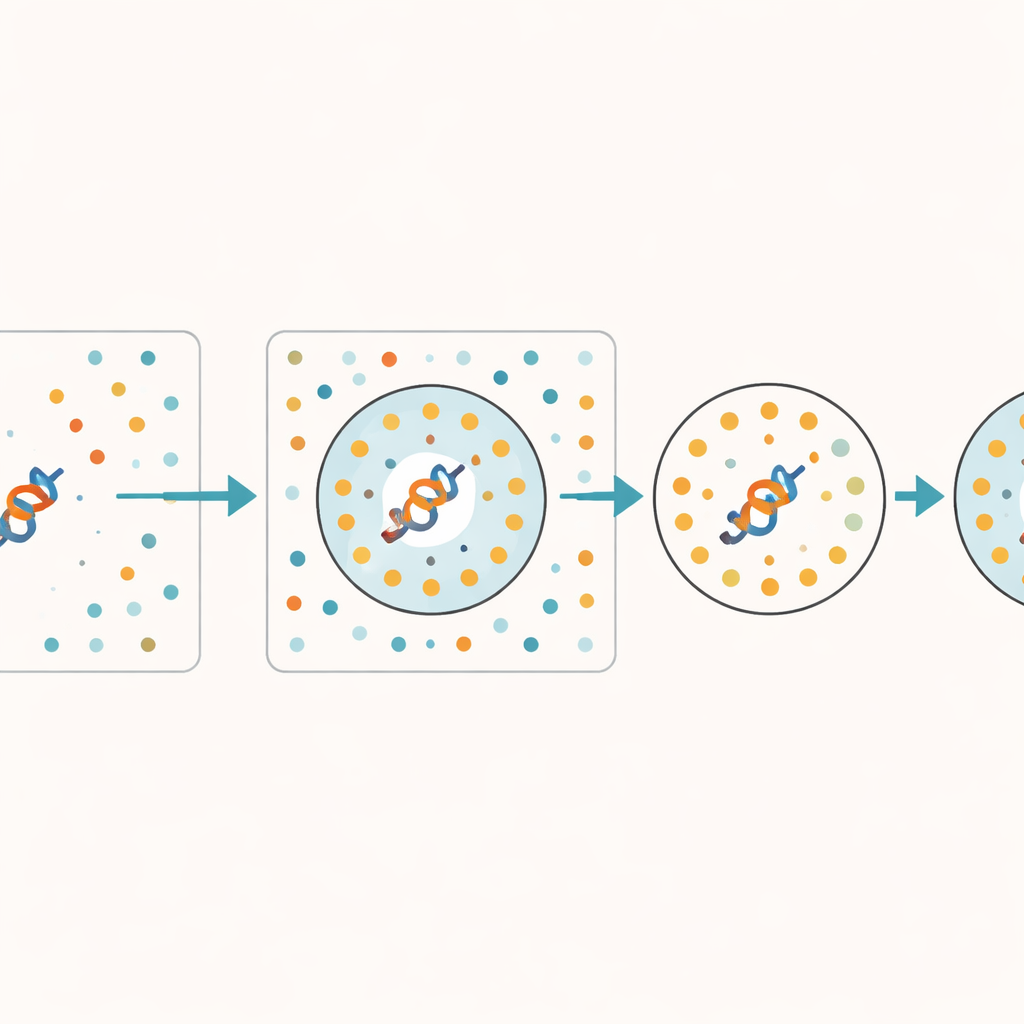
Les auteurs proposent une méthode pratique pour estimer la fiabilité d’un embedding sans modifier le modèle sous-jacent. Leur idée clé est de fournir au modèle un ensemble de séquences protéiques délibérément brouillées qui conservent la même composition de base mais perdent tous les motifs biologiquement significatifs. Ces séquences synthétiques servent de « casse » — une référence pour ce que le modèle produit lorsqu’il n’y a pas de véritable signal à apprendre. Pour chaque protéine réelle, la méthode vérifie combien de ses voisins les plus proches dans l’espace interne du modèle appartiennent à cette casse. Si de nombreux points proches proviennent de séquences brouillées, la représentation de la protéine est probablement peu apprise ou ambiguë. Les auteurs appellent cette fraction de voisins de la casse le Random Neighbor Score (RNS).
Relier l’incertitude à la performance réelle
Pour savoir si le RNS reflète réellement quelque chose d’important biologiquement, l’équipe a analysé de grandes collections de structures et de séquences protéiques en utilisant plusieurs modèles de pointe, dont ESM-2 et ProtT5. Ils ont constaté que les protéines dont la structure était bien prédite avaient tendance à présenter un faible RNS — ce qui signifie que leurs embeddings étaient éloignés de la casse. En revanche, les protéines avec de mauvaises prédictions structurelles se trouvaient dans des régions où séquences réelles et séquences brouillées se chevauchaient. Ce schéma se retrouvait à travers différents modèles et tâches. Lorsqu’ils ont examiné des problèmes plus appliqués, comme la prédiction des résidus en contact en 3D ou l’assignation de structures secondaires, ils ont observé une baisse claire de la précision lorsque le RNS augmentait. En d’autres termes, moins l’embedding est certain (RNS élevé), moins la prédiction en aval est fiable.
Zones d’ombre dans l’univers protéique
Le RNS a également révélé des angles morts systématiques dans la façon dont les modèles représentent différentes parties de l’univers protéique. Les régions intrinsèquement désordonnées — des segments flexibles qui n’ont pas de structure stable — présentaient de façon constante un RNS plus élevé que les domaines bien structurés, montrant que les modèles peinent davantage avec ces séquences glissantes. Même au sein du protéome humain bien étudié, une fraction substantielle de protéines avait un RNS non nul, indiquant qu’elles ne sont pas bien capturées par les modèles populaires. De manière surprenante, les modèles plus grands n’étaient pas toujours supérieurs : un grand modèle orienté structure pouvait être plus incertain sur de nombreuses protéines humaines qu’un modèle plus petit et plus général. Pour des protéines métagénomiques récemment découvertes et même pour des protéines « hallucinées » conçues par ordinateur pour paraître réalistes, un faible RNS suggérait que les modèles pouvaient généraliser avec confiance au-delà de leurs données d’entraînement lorsque les motifs sont cohérents.
De meilleurs filtres pour des insights biologiques supérieurs
Les auteurs ont ensuite testé comment le filtrage basé sur le RNS affecte une tâche cliniquement pertinente : prédire si un changement d’une seule lettre dans une protéine humaine est susceptible de perturber sa fonction ou de provoquer une maladie. En restreignant l’analyse aux protéines à faible RNS — où les embeddings semblaient fiables — les performances des modèles se sont nettement améliorées, atteignant souvent une forte discrimination entre variants nocifs et neutres. Pour les protéines à RNS élevé, les prédictions retombaient à des niveaux proches du hasard. Cela soutient l’idée que des embeddings peu fiables limitent silencieusement la meilleure précision possible de tout outil en aval construit sur eux, indépendamment des astuces d’entraînement.
Ce que cela implique pour l’utilisation de l’IA en biologie
Pour les non-spécialistes, la conclusion est que toutes les représentations protéiques dérivées par IA ne sont pas également dignes de confiance, et que cette fiabilité peut désormais être quantifiée. Le Random Neighbor Score agit comme un contrôle de santé simple et agnostique au modèle pour les embeddings : des scores faibles indiquent qu’une protéine se situe parmi d’autres séquences biologiquement significatives, tandis que des scores élevés suggèrent qu’elle dérive vers une casse de sosies aléatoires. En filtrant ou en pondérant les protéines en fonction du RNS avant de faire des prédictions structurelles, d’annoter des fonctions ou de prioriser des variants de maladie, les chercheurs peuvent se concentrer sur les régions où le modèle « comprend » réellement les données. Tout comme aucun scientifique n’utiliserait un microscope flou sans le remarquer, ce travail soutient que chaque modèle de langage protéique devrait être accompagné d’un moyen intégré d’évaluer la netteté de sa vision interne de la biologie.
Citation: Prabakaran, R., Bromberg, Y. Quantifying uncertainty in protein representations across models and tasks. Nat Methods 23, 796–804 (2026). https://doi.org/10.1038/s41592-026-03028-7
Mots-clés: modèles de langage protéique, fiabilité des embeddings, incertitude des représentations, prédiction des effets de variants, protéines intrinsèquement désordonnées