Clear Sky Science · pl
Kwantyfikacja niepewności reprezentacji białek w różnych modelach i zadaniach
Dlaczego wiarygodność AI dla białek ma znaczenie
Sztuczna inteligencja stała się potężnym mikroskopem dla niewidzialnego świata białek. Współczesne „modele języka białkowego” potrafią przewidzieć, jak białko wygląda w 3D i jak może się zachowywać, znając jedynie jego sekwencję cegiełek. Modele te już pomagają w projektowaniu nowych leków i zrozumieniu mutacji chorobotwórczych. Istnieje jednak ukryty problem: rzadko informują, na ile powinniśmy ufać wewnętrznym reprezentacjom, które generują. Artykuł wypełnia tę lukę, zadając proste pytanie o dalekosiężnych konsekwencjach: gdy model przekształca białko w chmurę liczb, jak rozpoznać, czy ta chmura naprawdę odzwierciedla biologię, czy jest jedynie szumem?
Od zdań do białek
Modele językowe zostały początkowo opracowane do przetwarzania tekstu ludzkiego, ucząc się relacji między słowami i przewidywania, co pojawi się dalej w zdaniu. Te same idee napędzają teraz modele czytające sekwencje białek i DNA jak długie słowa. Dla każdego białka model generuje „osadzenie” — punkt w wysokowymiarowej przestrzeni mający podsumować to, co model wie o danym białku. Te osadzenia są używane w wielu zadaniach końcowych, takich jak przewidywanie struktury, funkcji czy wpływu mutacji. Jednak w przeciwieństwie do znanych ocen predykcji czy miar pewności, osadzenia zwykle przyjmuje się bez zastrzeżeń: jeśli model zwraca wektor, użytkownicy mają tendencję ufać mu, nawet w obszarach przestrzeni białek, które model widział rzadko podczas treningu.
Rozpoznawanie, kiedy model zgaduje
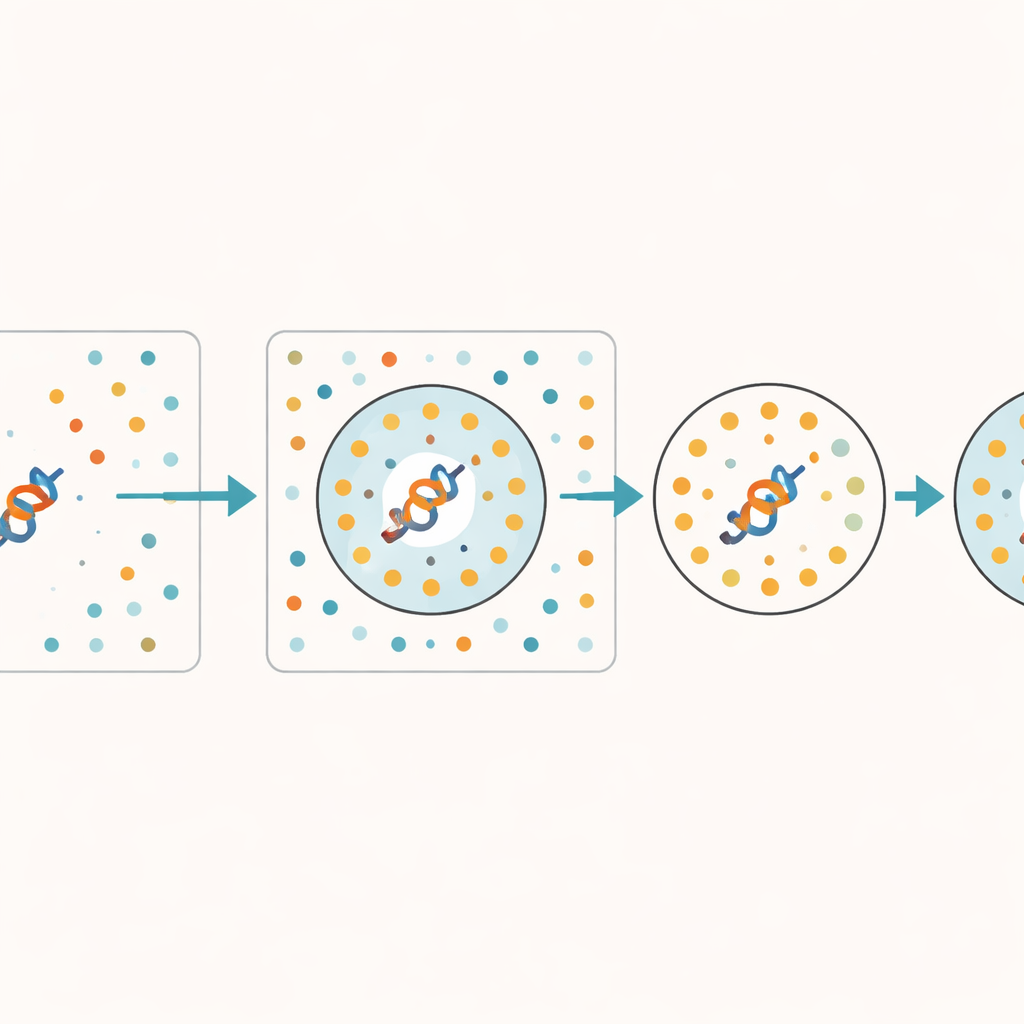
Autorzy proponują praktyczny sposób oszacowania, jak godne zaufania jest osadzenie, bez modyfikowania bazowego modelu. Kluczowy pomysł polega na podaniu modelowi zestawu celowo przemieszanych sekwencji białkowych, które zachowują podstawowy skład aminokwasowy, ale tracą wszystkie biologicznie znaczące wzorce. Te syntetyczne sekwencje służą jako „złomowisko” — punkt odniesienia dla tego, co model generuje, gdy nie ma prawdziwego sygnału do nauki. Dla każdego rzeczywistego białka metoda sprawdza, ile jego najbliższych sąsiadów w wewnętrznej przestrzeni modelu pochodzi z tego złomowiska. Jeśli wiele pobliskich punktów to sekwencje przemieszane, reprezentacja białka prawdopodobnie jest słabo wyuczona lub niejednoznaczna. Autorzy nazywają tę frakcję sąsiadów ze złomowiska Random Neighbor Score (RNS).
Powiązanie niepewności z wydajnością w praktyce
Aby sprawdzić, czy RNS rzeczywiście odzwierciedla coś biologicznie istotnego, zespół przeanalizował duże zbiory struktur i sekwencji białek przy użyciu kilku nowoczesnych modeli, w tym ESM-2 i ProtT5. Stwierdzili, że białka, których struktury przewidywano dokładnie, miały zwykle niski RNS — ich osadzenia były daleko od złomowiska. Natomiast białka ze słabymi przewidywaniami struktury leżały w rejonach, gdzie sekwencje rzeczywiste i przemieszane się pokrywały. Ten wzorzec utrzymywał się w różnych modelach i zadaniach. Gdy spojrzeli na bardziej zastosowane problemy, takie jak przewidywanie, które reszty aminokwasowe stykają się ze sobą w 3D czy przypisywanie struktury drugorzędowej, zaobserwowali wyraźny spadek dokładności wraz ze wzrostem RNS. Innymi słowy, im mniej pewne osadzenie (wyższy RNS), tym mniej wiarygodna predykcja oparta na nim.
Martwe pola w przestrzeni białek
RNS ujawnił też systematyczne martwe pola w sposobie reprezentowania różnych części wszechświata białkowego przez modele. Regiony wewnętrznie nieuporządkowane — elastyczne fragmenty pozbawione stabilnej struktury — miały konsekwentnie wyższy RNS niż dobrze ustrukturyzowane domeny, co pokazuje, że modele mają większe trudności z tymi śliskimi sekwencjami. Nawet w dobrze zbadanym proteomie człowieka znaczna część białek miała RNS różne od zera, co wskazuje, że nie są one dobrze uchwycone przez popularne modele. Co zaskakujące, większe modele nie zawsze były lepsze: duży, skoncentrowany na strukturze model mógł być bardziej niepewny wobec wielu białek ludzkich niż mniejszy, bardziej ogólny model. Dla nowo odkrytych białek metagenomicznych, a nawet komputerowo zaprojektowanych „halucynowanych” białek budowanych tak, by wyglądać realistycznie, niski RNS sugerował, że modele potrafią pewnie uogólnić poza dane treningowe, gdy wzorce są spójne.
Lepsze filtry dla lepszych wniosków biologicznych
Autorzy następnie przetestowali, jak filtrowanie oparte na RNS wpływa na zadanie o znaczeniu klinicznym: przewidywanie, czy pojedyncza zmiana literowa w białku człowieka prawdopodobnie zaburzy jego funkcję lub spowoduje chorobę. Gdy analizę ograniczono do białek o niskim RNS — gdzie osadzenia wydawały się wiarygodne — wydajność modeli poprawiła się znacząco, często osiągając silną separację między wariantami szkodliwymi a neutralnymi. Dla białek o wysokim RNS trafność predykcji spadała do poziomu rzutu monetą. To wspiera pogląd, że niewiarygodne osadzenia cicho ograniczają maksymalną możliwą dokładność dowolnego narzędzia opartego na nich, niezależnie od sprytnych trików treningowych.
Co to oznacza dla stosowania AI w biologii
Dla osób niebędących specjalistami wniosek jest prosty: nie wszystkie reprezentacje białek generowane przez AI są równie godne zaufania i tę wiarygodność można teraz zmierzyć. Random Neighbor Score działa jako proste, niezależne od modelu badanie kondycji osadzeń: niskie wartości wskazują, że białko znajduje się wśród innych biologicznie sensownych sekwencji, podczas gdy wysokie sugerują dryf w stronę złomowiska losowych podobieństw. Poprzez filtrowanie lub ważenie białek na podstawie RNS przed wykonywaniem przewidywań struktury, anotowaniem funkcji czy priorytetyzacją wariantów chorobowych, badacze mogą skupić się na obszarach, gdzie model rzeczywiście „rozumie” dane. Tak jak żaden naukowiec nie użyje nieostrego mikroskopu bez zwrócenia na to uwagi, ta praca argumentuje, że każdy model języka białkowego powinien mieć wbudowany sposób oceny ostrości swojego wewnętrznego obrazu biologii.
Cytowanie: Prabakaran, R., Bromberg, Y. Quantifying uncertainty in protein representations across models and tasks. Nat Methods 23, 796–804 (2026). https://doi.org/10.1038/s41592-026-03028-7
Słowa kluczowe: modele języka dla białek, wiarygodność osadzeń, niepewność reprezentacji, predykcja efektu wariantów, białka wewnętrznie nieuporządkowane