Clear Sky Science · zh
利用全局相互作用改进费米子量子模拟策略
为何加速量子化学至关重要
设计新药、更好电池和先进材料,往往取决于理解电子在分子内部如何运动与相互作用。当经典超级计算机尝试精确跟踪这些多电子系统时,很快就力不从心。囚禁离子量子计算机被视为一种前进的途径,但目前它们的计算仍然缓慢且受噪声影响。本文展示了如何利用囚禁离子的天然优势,以远少于以往的操作次数运行一类关键的化学计算,从而将精确的量子模拟更进一步地推向可行现实。
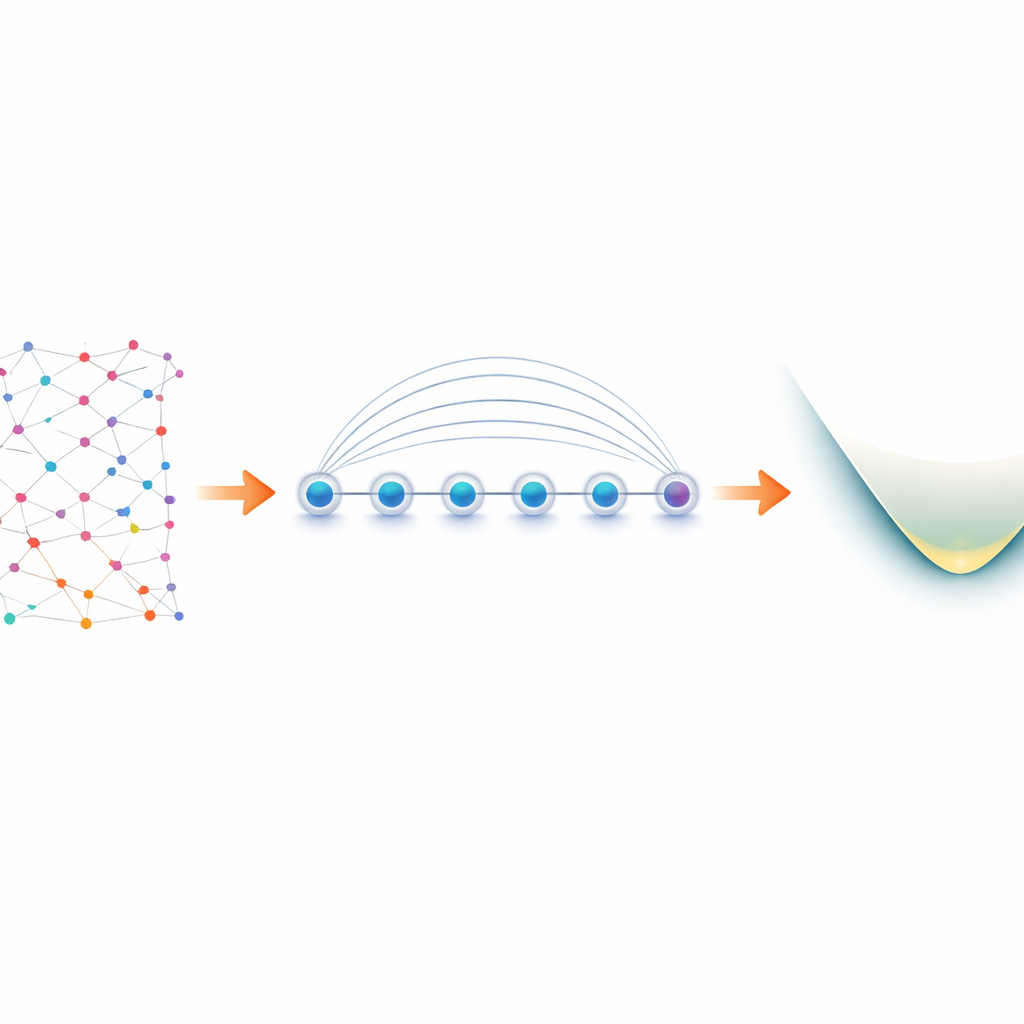
从分子中的电子到实验室里的量子比特
分子中的电子表现为“费米子”,遵循关于能否共享量子态的严格规则。为了在量子计算机上模拟它们,研究者首先将这些费米子规则翻译为作用于量子比特的操作,这一过程称为映射。一个流行的选择是 Jordan–Wigner 映射,其概念上简单但会产生长程相互作用:对电子的一个逻辑操作可能变成跨越整个器件的一串耦合量子比特。在大多数只允许相邻量子比特直接相互作用的量子硬件上,这会导致电路膨胀并产生额外且易出错的交换操作。囚禁离子器件有所不同。排列在线上的离子可以利用一种本征操作——Mølmer–Sørensen(MS)门——一次性产生纠缠,天然地连接远端量子比特。作者们正是利用这种全局相互作用,将 Jordan–Wigner 的表面弱点转化为优势。
将全局相互作用用作捷径
许多化学算法的核心是“激发算符”,它描述了电子从占据轨道跃迁到空轨道的过程。这些算符出现在两个主要场合:用于寻找分子基态的幺正耦合簇(UCC)方法,以及用于逐步(Trotter 化)模拟电子系统随时间演化的步骤。先前在囚禁离子机器上的方案对激发算符的每一部分分别实现,对每一项使用多个 MS 门。在这项工作中,作者展示了特定形式的 MS 门可以一次对整类这些部分进行对角化。通过在仅两个 MS 门之间放置简单的单量子比特旋转,他们能够并行应用许多非局域分量。对于单电子激发,这将所需的 MS 门数减半;对于双电子激发,则将其减少到四分之一,而不需要任何额外的辅助量子比特。
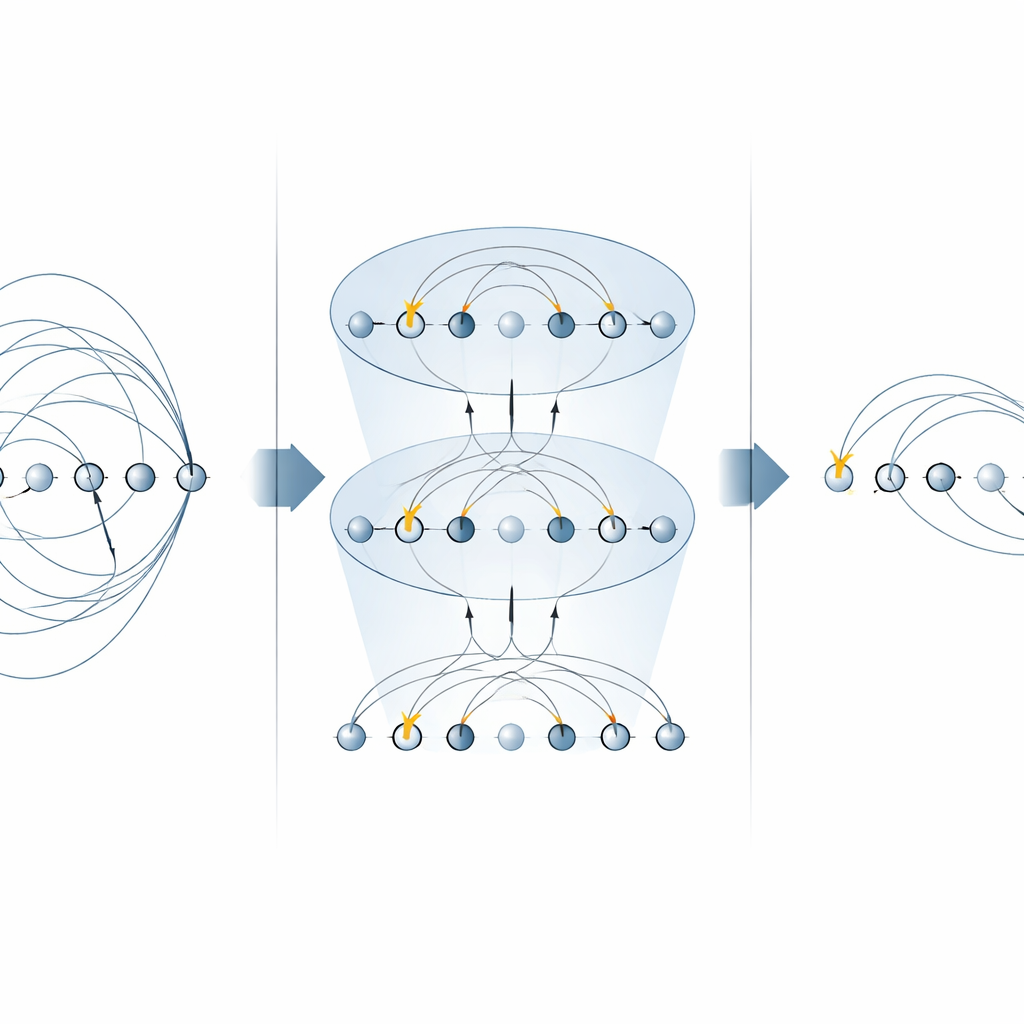
构建更快的量子化学电路
利用这些优化的构件,作者构建了完整的电路,用于变分基态搜索和实时动力学两种任务。他们在一个小但非平凡的分子离子 H3+ 上演示了该方法,展示如何用远少于早期方法的全局门组装整个 UCCSD(单激发与双激发)层以及用于时间演化的 Trotter 步。相同策略可推广到更高阶的激发,对近中期设备流行的替代“量子比特激发”猜设也有益,并且可复用来直接模拟电子哈密顿量。重要的是,该方法保持了粒子数与自旋等关键物理量的守恒,这些在化学应用中至关重要。
在现实噪声下测试性能
电路更短只有在实际硬件上确实降低错误时才有意义。为此,团队构建了一个针对 12 离子线性阱的详细噪声模型,包括振动模式频率和激光功率的波动——这些是当今实验中的主要误差源。然后他们将新电路与标准电路在一系列小分子上进行了比较,追踪能量误差、量子保真度损失以及守恒量的违背情况。在单、双激发范围内,改进的设计将电路不保真度降低了大约一半到一个数量级。在完整的分子计算中,它们始终使模拟能量和物理观测值更接近理想结果,并且对于更复杂激发和更大系统,其优势越发明显。
这对未来模拟意味着什么
该研究并不主张完美的量子化学已经近在咫尺;在当前噪声水平下,即使是改进后的电路常常也难以单独超越最佳的经典近似。然而,这项工作表明,通过将算法与硬件相匹配——在这里是使费米子激发结构与全局离子阱相互作用对齐——可以显著减少开销并提高精度。结合误差缓解技术以及诸如基于量子比特的激发等较为温和的近似,这些策略可能使近中期的囚禁离子设备能够处理那些刚好超出经典计算机可及范围的化学相关问题。
引用: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
关键词: 囚禁离子量子计算, 费米子模拟, 分子量子化学, Mølmer–Sørensen 门, 幺正耦合簇