Clear Sky Science · pl
Ulepszone strategie symulacji fermionowej na komputerach kwantowych z interakcjami globalnymi
Dlaczego przyspieszenie chemii kwantowej ma znaczenie
Projektowanie nowych leków, lepszych baterii i zaawansowanych materiałów często sprowadza się do zrozumienia, jak elektrony poruszają się i oddziałują wewnątrz cząsteczek. Klasyczne superkomputery szybko tracą wydajność, gdy próbują śledzić takie wieloelektronowe układy dokładnie. Komputery kwantowe oparte na jonach zamkniętych w pułapkach obiecują drogę naprzód, ale dziś ich obliczenia są wciąż powolne i podatne na szumy. Artykuł pokazuje, jak wykorzystać naturalne zalety układów z jonami pułapkowymi, by wykonać istotną klasę obliczeń chemicznych przy znacznie mniejszej liczbie operacji, przybliżając dokładne symulacje kwantowe do praktycznej użyteczności.
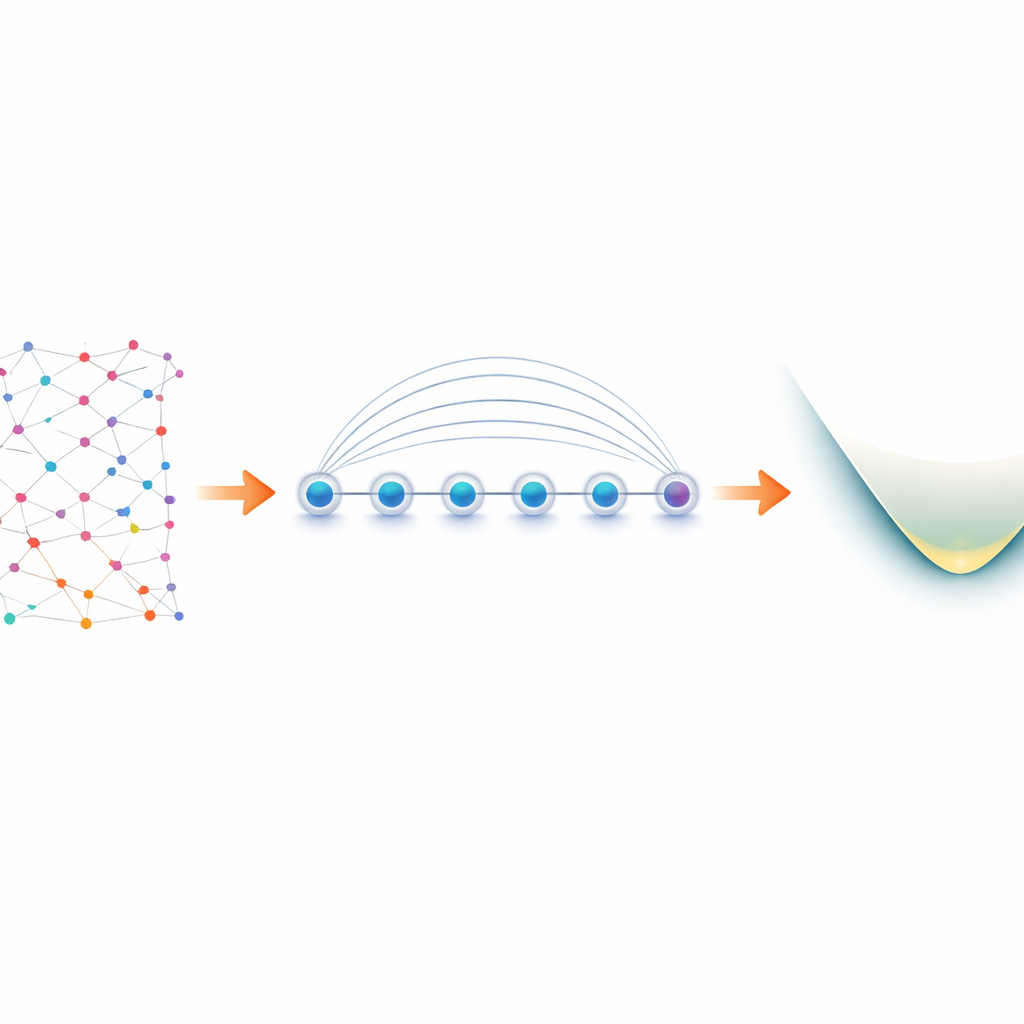
Od elektronów w cząsteczce do kubitów w laboratorium
Elektrony w cząsteczce zachowują się jako „fermiony”, podlegając ścisłym regułom dotyczących tego, jak mogą dzielić stany kwantowe. Aby symulować je na komputerze kwantowym, badacze najpierw przekształcają te fermionowe reguły w operacje na kubitach — proces zwany mapowaniem. Popularny wybór, mapowanie Jordana–Wignera, jest koncepcyjnie prosty, ale generuje oddziaływania dalekiego zasięgu: jedna logiczna operacja na elektronach może stać się łańcuchem sprzężonych kubitów rozciągniętym na całe urządzenie. Na większości platform kwantowych, które pozwalają na bezpośrednią komunikację tylko sąsiednich kubitów, prowadzi to do zwiększenia obwodów i dodatkowych, podatnych na błędy, operacji swap. Urządzenia z jonami pułapkowymi są inne. Jony ustawione w linii można zaplątać jednocześnie za pomocą natywnej operacji zwanej bramką Mølmer–Sørensen (MS), która naturalnie łączy oddalone kubity. Autorzy wykorzystują tę globalną interakcję, by przemienić pozorną słabość mapowania Jordana–Wignera w siłę.
Wykorzystanie interakcji globalnych jako skrótu
Rdzeniem wielu algorytmów chemicznych jest „operator wzbudzeń”, opisujący przesuwanie elektronów z zajętych orbitalów do pustych. Operatory te występują w dwóch głównych miejscach: w metodzie unitary coupled‑cluster (UCC) do znajdowania stanów podstawowych cząsteczek oraz w krokowych (trotteryzowanych) symulacjach ewolucji układu elektronicznego w czasie. Wcześniejsze schematy na maszynach z jonami pułapkowymi realizowały każdy składnik operatora wzbudzeń oddzielnie, używając wielu bramek MS na każdy termin. W tej pracy autorzy pokazują, że specyficzne formy bramki MS mogą zdiagnoalizować całe rodziny tych składników jednocześnie. Umieszczając proste rotacje pojedynczych kubitów pomiędzy tylko dwiema bramkami MS, potrafią zastosować wiele nielokalnych składników równolegle. Dla wzbudzeń pojedynczoelektronowych zmniejsza to liczbę potrzebnych bramek MS o połowę, a dla wzbudzeń dwu‑elektronowych — aż czterokrotnie, bez konieczności używania dodatkowych kubitów pomocniczych.
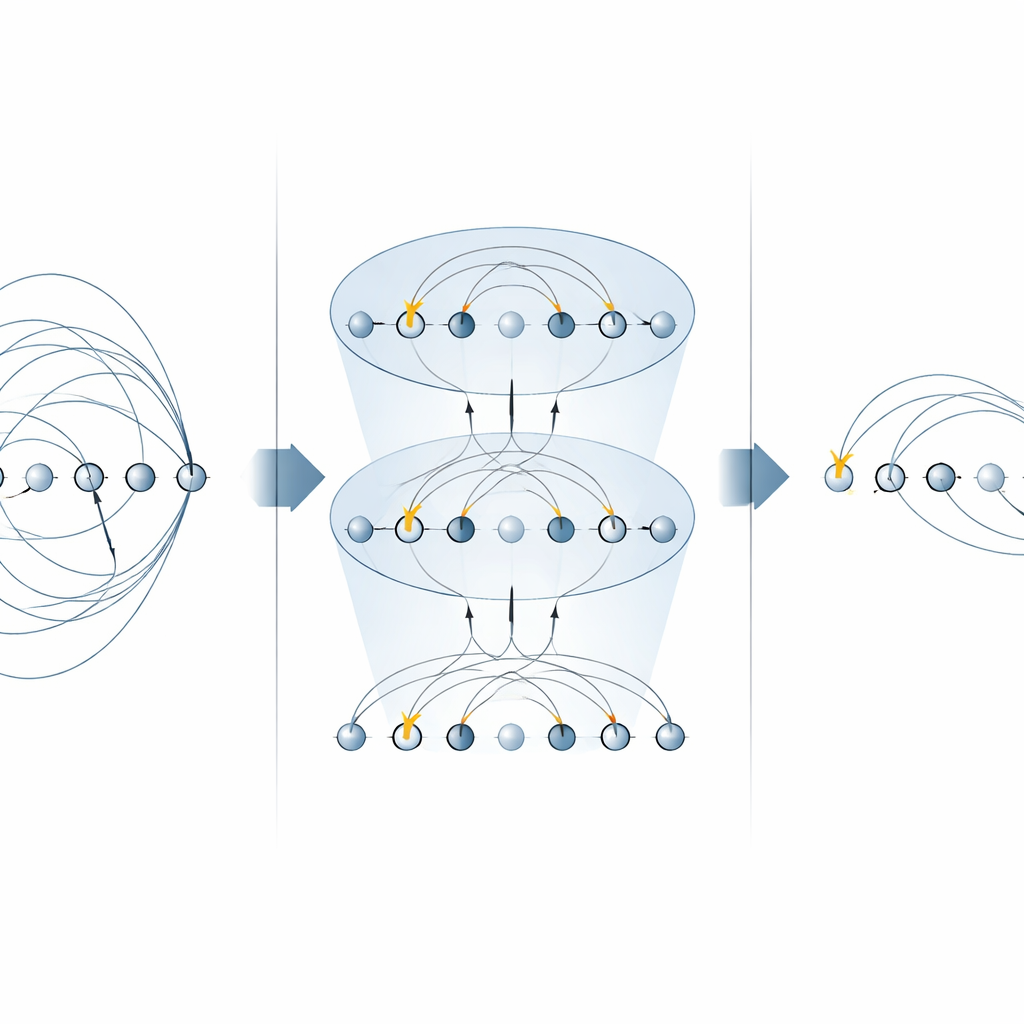
Budowanie szybszych obwodów do chemii kwantowej
Dzięki tym zoptymalizowanym blokom konstrukcyjnym autorzy budują kompletne obwody zarówno do wariacyjnego poszukiwania stanu podstawowego, jak i do ewolucji w czasie rzeczywistym. Ilustrują metodę na małym, lecz nietrywialnym jonie molekularnym H3+, pokazując, jak złożyć całą warstwę UCCSD (singles‑and‑doubles) oraz krok Trottera dla ewolucji czasowej, używając znacznie mniejszej liczby bramek globalnych niż w podejściach wcześniejszych. Ta sama strategia uogólnia się na wzbudzenia wyższych rzędów, przynosi korzyści alternatywnym ansatzom „wzbudzeń kubitów” popularnym w urządzeniach bliskoterminowych i może być ponownie wykorzystana do bezpośredniej symulacji hamitonianów elektronicznych. Co ważne, podejście zachowuje kluczowe wielkości fizyczne, takie jak liczba cząstek i spin, które są centralne dla zastosowań w chemii.
Testowanie wydajności w warunkach realistycznego szumu
Krótsze obwody pomagają tylko wtedy, gdy rzeczywiście zmniejszają błędy na prawdziwym sprzęcie. Aby to sprawdzić, zespół zbudował szczegółowy model szumów liniowej pułapki na 12 jonów, uwzględniając fluktuacje częstotliwości trybów drgań i mocy laserów — główne źródła błędów w dzisiejszych eksperymentach. Następnie porównali swoje nowe obwody ze standardowymi dla szeregu małych cząsteczek, śledząc błędy energii, utratę wierności kwantowej i naruszenia zachowanych wielkości. Zarówno dla wzbudzeń pojedynczych, jak i podwójnych, ulepszone projekty zmniejszyły infidelność obwodu w przybliżeniu o połowę do rzędu wielkości. W pełnych obliczeniach molekularnych konsekwentnie przybliżały energie symulowane i obserwowalne fizyczne do wyników idealnych, a ich przewaga stawała się bardziej widoczna dla bardziej złożonych wzbudzeń i większych układów.
Co to znaczy dla przyszłych symulacji
Badanie nie twierdzi, że perfekcyjna chemia kwantowa jest tuż za rogiem; przy obecnych poziomach szumów nawet ulepszone obwody często nie wystarczają, by same przewyższyć najlepsze klasyczne przybliżenia. Jednak praca pokazuje, że dopasowując algorytmy do sprzętu — w tym przypadku wyrównując strukturę fermionowych wzbudzeń z globalnymi interakcjami w pułapce jonowej — można dramatycznie zmniejszyć narzut i poprawić dokładność. W połączeniu z technikami łagodzenia błędów i bardziej umiarkowanymi przybliżeniami, takimi jak wzbudzenia oparte na kubitach, te strategie mogą umożliwić urządzeniom z jonami pułapkowymi rozwiązanie chemicznie istotnych problemów, które są nieco poza zasięgiem klasycznych komputerów.
Cytowanie: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Słowa kluczowe: komputery kwantowe z jonami pułapkowymi, symulacja fermionowa, kwantowa chemia molekularna, bramki Mølmer–Sørensen, unitary coupled cluster