Clear Sky Science · de
Verbesserte Strategien für fermionische Quanten‑Simulationen mit globalen Wechselwirkungen
Warum die Beschleunigung der Quantenchemie wichtig ist
Die Entwicklung neuer Medikamente, besserer Batterien und fortschrittlicher Materialien hängt oft davon ab, zu verstehen, wie sich Elektronen innerhalb von Molekülen bewegen und miteinander wechselwirken. Klassische Supercomputer stoßen schnell an ihre Grenzen, wenn sie diese Viel‑Elektronen‑Systeme exakt verfolgen sollen. Quantencomputer mit gefangenen Ionen versprechen einen Ausweg, doch ihre Berechnungen sind heute noch langsam und verrauscht. Diese Arbeit zeigt, wie man die natürlichen Stärken von Ionenfallen nutzt, um eine wichtige Klasse von Chemie‑Berechnungen mit deutlich weniger Operationen durchzuführen und damit genaue Quantensimulationen einen Schritt näher an die praktische Anwendbarkeit zu bringen.
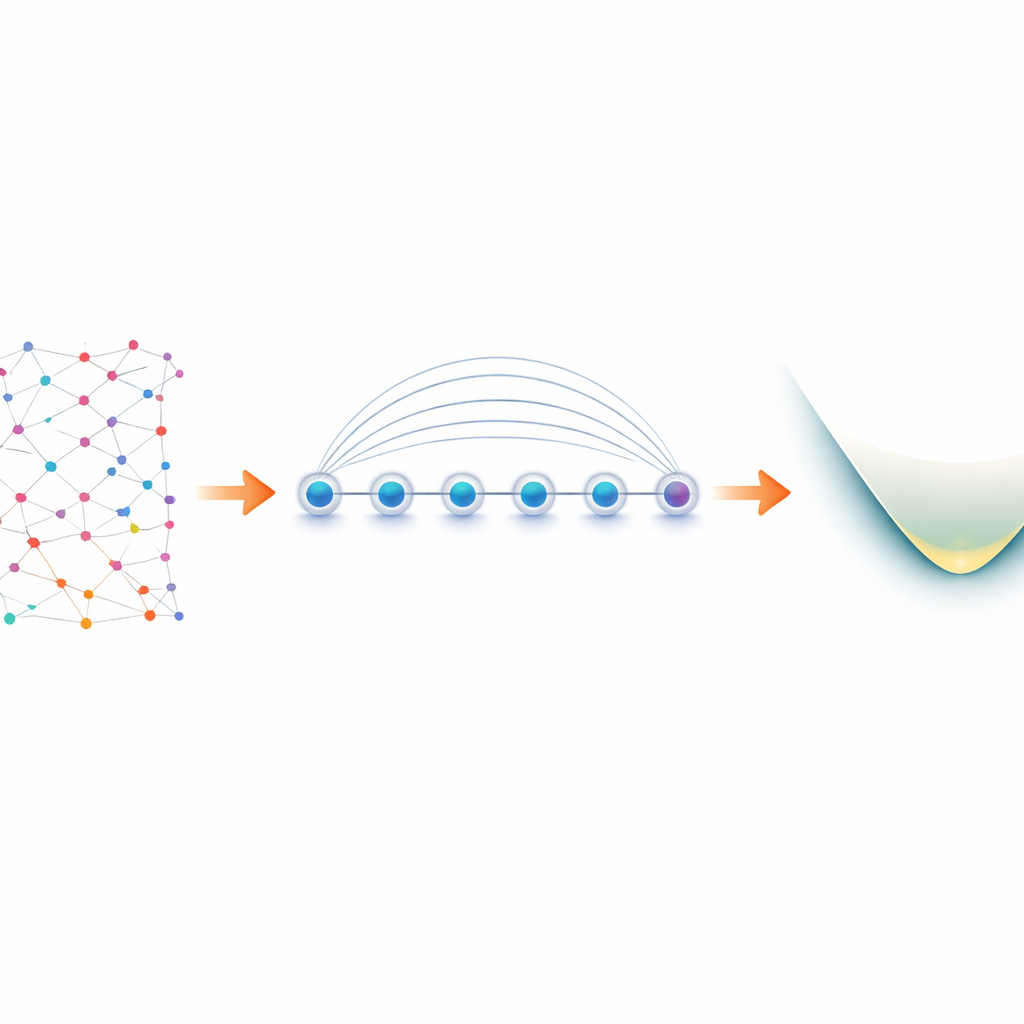
Von Elektronen in Molekülen zu Qubits im Labor
Elektronen in einem Molekül verhalten sich als „Fermionen“ und unterliegen strikten Regeln, wie sie Quantenzustände teilen können. Um sie auf einem Quantencomputer zu simulieren, übersetzen Forschende diese fermionischen Regeln zuerst in Operationen auf Qubits, ein Prozess, der als Mapping bekannt ist. Eine gängige Wahl, das Jordan–Wigner‑Mapping, ist konzeptionell einfach, erzeugt aber langreichweitige Wechselwirkungen: Eine logische Operation auf den Elektronen kann zu einer Kette gekoppelter Qubits werden, die sich über das ganze Gerät erstreckt. Auf den meisten Quantenhardware‑Plattformen, die nur direkte Wechselwirkung benachbarter Qubits erlauben, führt das zu aufgeblähten Schaltkreisen und zusätzlichen fehleranfälligen Swap‑Operationen. Geräte mit gefangenen Ionen sind anders. In einer Linie angeordnete Ionen können gleichzeitig mit einer nativen Operation, dem Mølmer–Sørensen (MS)‑Gatter, verschränkt werden, das entfernte Qubits natürlich verbindet. Die Autorinnen und Autoren nutzen diese globale Wechselwirkung, um die vermeintliche Schwäche des Jordan–Wigner‑Mappings in eine Stärke zu verwandeln.
Globale Wechselwirkungen als Abkürzung nutzen
Im Kern vieler Chemie‑Algorithmen steht der „Anregungsoperator“, der beschreibt, wie Elektronen von besetzten in unbesetzte Orbitale bewegt werden. Diese Operatoren treten an zwei wichtigen Stellen auf: in der unitary coupled‑cluster (UCC)‑Methode zur Bestimmung von Molekülgrundzuständen und in schrittweisen (trotterisierten) Simulationen der zeitlichen Entwicklung eines elektronischen Systems. Frühere Ansätze auf Ionenfallen‑Maschinen setzten jeden Teil eines Anregungsoperators separat um und verwendeten dabei mehrere MS‑Gatter für jeden Term. In dieser Arbeit zeigen die Autorinnen und Autoren, dass spezielle Formen des MS‑Gatters ganze Familien dieser Terme gleichzeitig diagonalisieren können. Indem sie einfache Einzel‑Qubit‑Rotationen nur zwischen zwei MS‑Gattern platzieren, können sie viele nicht‑lokale Komponenten parallel anwenden. Bei Ein‑Elektron‑Anregungen halbiert dies die Anzahl der benötigten MS‑Gatter, bei Zwei‑Elektron‑Anregungen reduziert es sie um den Faktor vier, ohne zusätzliche Hilfsqubits zu benötigen.
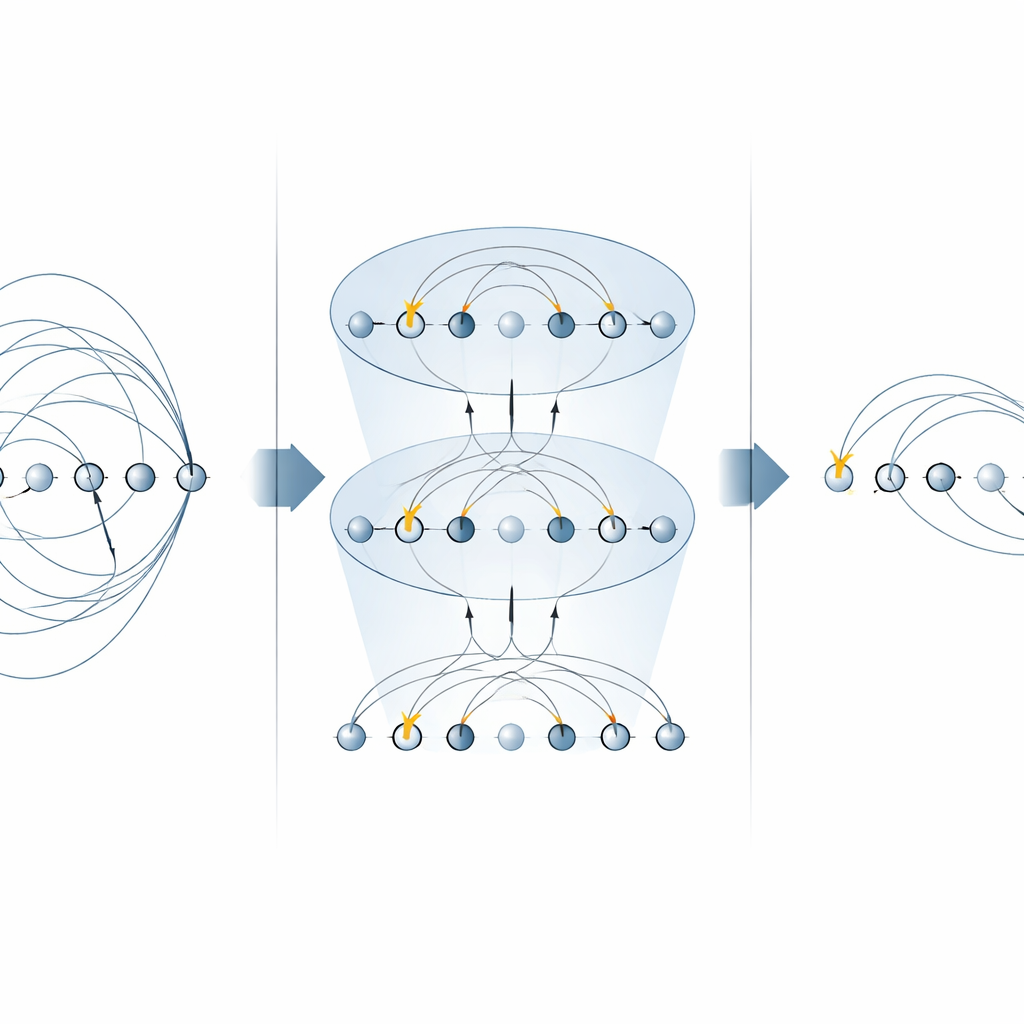
Schnellere Quantenchemie‑Schaltkreise aufbauen
Mit diesen optimierten Bausteinen bauen die Autorinnen und Autoren komplette Schaltkreise sowohl für variationale Grundzustandssuchen als auch für Echtzeitdynamiken. Sie illustrieren die Methode an einem kleinen, aber nichttrivialen Molekülion, H3+, und zeigen, wie sich eine gesamte UCCSD‑(Singles‑und‑Doubles)‑Schicht sowie ein Trotter‑Schritt für die Zeitentwicklung mit deutlich weniger globalen Gattern als in früheren Ansätzen zusammensetzen lassen. Dieselbe Strategie verallgemeinert sich auf höhergradige Anregungen, bringt Vorteile für alternative „Qubit‑Anregungs“‑Ansätze, die bei nahterm‑tauglichen Geräten beliebt sind, und kann wiederverwendet werden, um elektronische Hamiltonoperatoren direkt zu simulieren. Wichtig ist, dass der Ansatz zentrale physikalische Größen wie Teilchenzahl und Spin respektiert, die in der Chemie von zentraler Bedeutung sind.
Leistung unter realistischem Rauschen testen
Kürzere Schaltkreise helfen nur, wenn sie auf echter Hardware tatsächlich Fehler reduzieren. Um dies zu prüfen, erstellte das Team ein detailliertes Rauschmodell für eine 12‑Ionen‑Linienfalle, das Fluktuationen in Schwingungsmodenfrequenzen und Laserleistungen einschließt — wichtige Fehlerquellen in heutigen Experimenten. Anschließend verglichen sie ihre neuen Schaltkreise mit Standardvarianten für eine Reihe kleiner Moleküle und verfolgten Energiefehler, Verlust an Quanten‑Fidelity und Verletzungen von Erhaltungsgrößen. Bei Einzel‑ und Doppelanregungen verringerten die verbesserten Entwürfe die Schaltkreis‑Infidelity um etwa die Hälfte bis zu einer Größenordnung. Für vollständige Molekülrechnungen brachten sie konsistent simulierte Energien und physikalische Observablen näher an ideale Ergebnisse, und ihr Vorteil wurde bei komplexeren Anregungen und größeren Systemen stärker ausgeprägt.
Was das für zukünftige Simulationen bedeutet
Die Studie behauptet nicht, dass perfekte Quantenchemie unmittelbar bevorsteht; bei den aktuellen Rauschpegeln liegen selbst die verbesserten Schaltkreise oft noch unterhalb der besten klassischen Näherungen. Doch die Arbeit zeigt, dass durch das Abstimmen von Algorithmen auf die Hardware — in diesem Fall die Angleichung der Struktur fermionischer Anregungen an globale Ion‑Trap‑Wechselwirkungen — der Overhead drastisch reduziert und die Genauigkeit verbessert werden kann. In Kombination mit Fehler‑Mitigationstechniken und moderateren Näherungen wie qubit‑basierten Anregungen könnten diese Strategien es ermöglichen, dass nahterm‑taugliche Ionenfallen‑Geräte chemisch relevante Probleme lösen, die für klassische Computer derzeit knapp unerreichbar sind.
Zitation: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Schlüsselwörter: Quantencomputing mit gefangenen Ionen, fermionische Simulation, molekulare Quantenchemie, Mølmer–Sørensen‑Gatter, unitary coupled cluster