Clear Sky Science · nl
Verbeterde strategieën voor fermionische kwantumsimulatie met globale interacties
Waarom het versnellen van kwantumchemie ertoe doet
Het ontwerpen van nieuwe geneesmiddelen, betere batterijen en geavanceerde materialen komt vaak neer op het begrijpen hoe elektronen zich bewegen en met elkaar wisselwerken in moleculen. Klassieke supercomputers lopen snel tegen hun grenzen aan wanneer ze deze veel‑elektronensystemen exact proberen bij te houden. Gevangen‑ion kwantumcomputers bieden een uitweg, maar op dit moment zijn hun berekeningen nog traag en ruisgevoelig. Dit artikel laat zien hoe de natuurlijke sterke punten van gevangen ionen benut kunnen worden om een belangrijke klasse chemische berekeningen met veel minder bewerkingen uit te voeren, en brengt accurate kwantumsimulaties een stap dichter bij praktische toepassing.
Van elektronen in moleculen naar qubits in het lab
Elektronen in een molecuul gedragen zich als “fermionen” en volgen strikte regels over hoe zij kwantumtoestanden kunnen delen. Om ze op een kwantumcomputer te simuleren, vertalen onderzoekers deze fermionische regels eerst naar operaties op qubits, een proces dat bekendstaat als mapping. Een populaire keuze, de Jordan–Wigner‑mapping, is conceptueel eenvoudig maar genereert langafstandsinteracties: één logische operatie op elektronen kan een string van gekoppelde qubits worden die over het hele apparaat is uitgestrekt. Op de meeste kwantumhardware, die alleen buren direct met elkaar laat communiceren, leidt dit tot opgeblazen circuits en foutgevoelige extra swap‑operaties. Gevangen‑ion apparaten zijn anders. Ionen in een lijn kunnen in één keer verstrengeld worden met een native operatie genaamd de Mølmer–Sørensen (MS)-poort, die van nature verre qubits verbindt. De auteurs benutten deze globale interactie om de schijnbare zwakte van Jordan–Wigner om te zetten in een kracht.
Globale interacties als snelkoppeling gebruiken
De kern van veel chemiealgoritmen is de “excitatoperator”, die beschrijft hoe elektronen verplaatst worden van bezette naar lege orbitalen. Deze operatoren verschijnen op twee belangrijke plaatsen: de unitary coupled‑cluster (UCC)-methode om moleculaire grondtoestanden te vinden, en in stap‑voor‑stap (Trotteriseerde) simulaties van hoe een elektronisch systeem in de tijd evolueert. Eerdere schema’s op gevangen‑ion machines implementeerden elk deel van een excitatieoperator afzonderlijk, waarbij meerdere MS‑poorten voor elk term gebruikt werden. In dit werk laten de auteurs zien dat specifieke vormen van de MS‑poort hele families van deze termen tegelijk kunnen diagonaleren. Door eenvoudige single‑qubit rotaties tussen slechts twee MS‑poorten te plaatsen, kunnen ze veel niet‑lokale componenten parallel toepassen. Voor enkelvoudige elektronenexcitaties halveert dit het aantal vereiste MS‑poorten, en voor dubbel‑elektronexcitaties vermindert het ze met een factor vier, zonder extra hulpqubits nodig te hebben.
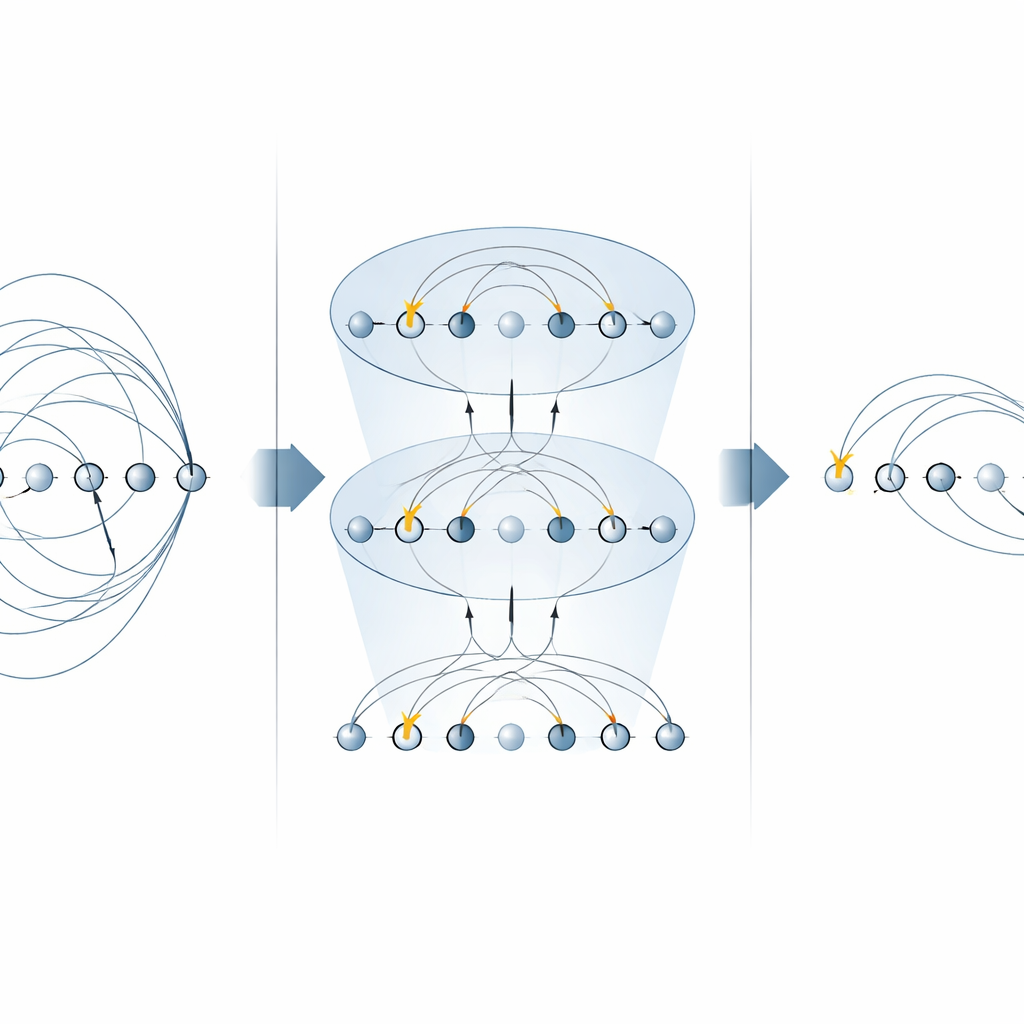
Snellere kwantumchemiecircuits opbouwen
Met deze geoptimaliseerde bouwblokken construeren de auteurs complete circuits voor zowel variationale grondtoestandzoektochten als realtime dynamica. Ze illustreren de methode op een klein maar niet‑triviaal moleculair ion, H3+, en tonen hoe een volledige UCCSD‑laag (singles‑en‑doubles) en een Trotter‑stap voor tijdsevolutie opgebouwd kunnen worden met veel minder globale poorten dan eerdere benaderingen. Dezelfde strategie is te generaliseren naar hogere‑orde excitatiess, brengt voordelen voor alternatieve “qubit‑excitaties” ansätze die populair zijn voor kortetermijnapparaten, en kan hergebruikt worden voor het rechtstreeks simuleren van elektronische Hamiltonianen. Belangrijk is dat de aanpak essentiële fysieke grootheden zoals de deeltjesaantallen en spin respecteert, die centraal staan in chemietoepassingen.
Prestaties testen onder realistische ruis
Kortere circuits helpen alleen als ze daadwerkelijk fouten op echte hardware verminderen. Om dit te controleren bouwde het team een gedetailleerd ruismodel van een lineaire val van 12 ionen, inclusief fluctuaties in de frequenties van trillingstoestanden en laserkrachten — belangrijke foutbronnen in huidige experimenten. Vervolgens vergeleken ze hun nieuwe circuits met standaardontwerpen voor een reeks kleine moleculen, en volgden energiefouten, verlies aan kwantumfideliteit en schendingen van bewaarde grootheden. Over enkel‑ en dubbelexcitaties verminderden de verbeterde ontwerpen de circuit‑infideliteit ruwweg met de helft tot een volledige orde van grootte. Voor volledige moleculaire berekeningen brachten ze consequent gesimuleerde energieën en fysische waarnemingen dichter bij ideale resultaten, en hun voordeel werd duidelijker bij complexere excitatiess en grotere systemen.
Wat dit betekent voor toekomstige simulaties
De studie beweert niet dat perfecte kwantumchemie om de hoek ligt; bij de huidige ruisniveaus schieten zelfs de verbeterde circuits vaak tekort om op zichzelf de beste klassieke benaderingen te overtreffen. Het werk toont echter aan dat door algoritmen op de hardware af te stemmen — in dit geval het uitlijnen van fermionische excitatiestructuur met globale ion‑val interacties — de overhead dramatisch kan worden verminderd en de nauwkeurigheid kan verbeteren. Gecombineerd met fout‑mitigatietechnieken en meer bescheiden benaderingen zoals qubit‑gebaseerde excitaties, zouden deze strategieën kortetermijn gevangen‑ion apparaten in staat kunnen stellen chemisch relevante problemen aan te pakken die net buiten het bereik van klassieke computers liggen.
Bronvermelding: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Trefwoorden: gevangen ion kwantumcomputing, fermionische simulatie, moleculaire kwantumchemie, Mølmer–Sørensen-poorten, unitary coupled cluster