Clear Sky Science · sv
Förbättrade strategier för fermionisk kvantsimulering med globala interaktioner
Varför det spelar roll att snabba upp kvantkemi
Att designa nya läkemedel, bättre batterier och avancerade material handlar ofta om att förstå hur elektroner rör sig och interagerar i molekyler. Klassiska superdatorer får snabbt svårt att exakt följa dessa mångelektronsystem. Fångade‑jon‑kvantdatorer lovar en väg framåt, men idag är deras beräkningar fortfarande långsamma och brusiga. Denna artikel visar hur man kan utnyttja fångade joners naturliga styrkor för att köra en viktig klass kemiberäkningar med avsevärt färre operationer, vilket för flyttbara kvantsimuleringar ett steg närmare praktisk verklighet.
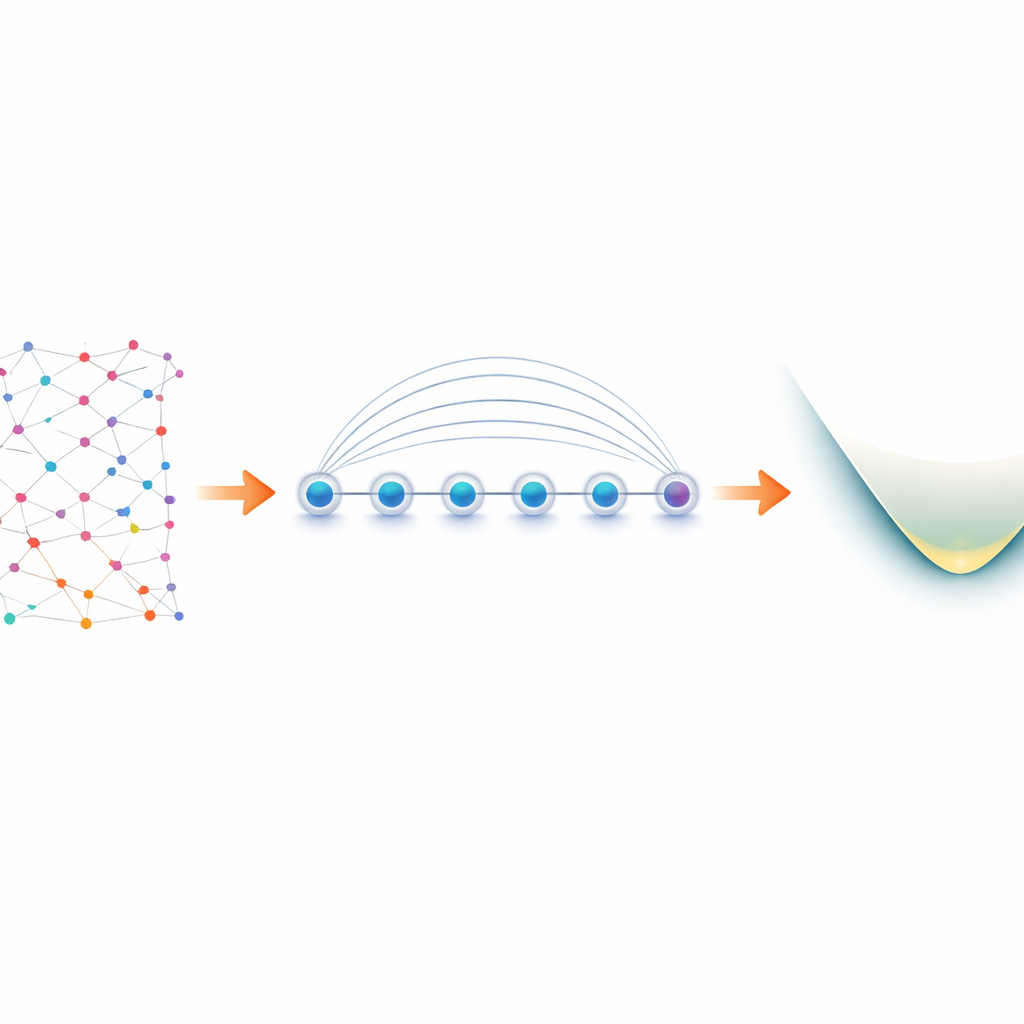
Från elektroner i molekyler till qubitar i laboratoriet
Elektroner i en molekyl beter sig som ”fermioner” och följer strikta regler för hur de kan dela kvanttillstånd. För att simulera dem på en kvantdator översätter forskare först dessa fermionregler till operationer på qubitar, en process som kallas mappning. Ett populärt val, Jordan–Wigner‑mappningen, är konceptuellt enkel men ger upphov till långräckviddsinteraktioner: en logisk elektronstruktur kan bli en kedja av ihopkopplade qubitar som sträcker sig över hela enheten. På de flesta kvantplattformar, som bara tillåter närliggande qubitar att kommunicera direkt, leder detta till stora kretsar och extra felfyllda swap‑operationer. Fångade‑jonsenheter är annorlunda. Joner ordnade i en rad kan sammanflätas samtidigt med en inbyggd operation kallad Mølmer–Sørensen (MS)‑grind, som naturligt kopplar avlägsna qubitar. Författarna utnyttjar denna globala interaktion för att vända Jordans–Wigners till synes svaghet till en fördel.
Att använda globala interaktioner som genväg
Kärnan i många kemialgoritmer är ”excitationsoperatorn”, som beskriver att flytta elektroner från besatta till tomma orbitaler. Dessa operatorer dyker upp på två huvudställen: den unitära kopplade‑kluster‑metoden (UCC) för att hitta molekylära grundtillstånd, och i stegvisa (Trotteriserade) simuleringar av hur ett elektroniskt system utvecklas i tiden. Tidigare scheman på fångade‑jonsmaskiner implementerade varje del av en excitationsoperator separat och använde flera MS‑grindar för varje term. I detta arbete visar författarna att specifika former av MS‑grinden kan diagonaliser hela familjer av dessa termer på en gång. Genom att placera enkla enkla‑qubitrotationer mellan bara två MS‑grindar kan de tillämpa många icke‑lokala komponenter parallellt. För enkla‑electronexcitationer halverar detta antalet nödvändiga MS‑grindar, och för dubbel‑electronexcitationer minskar det antalet med en faktor fyra, utan att behöva extra hjälparqubit.
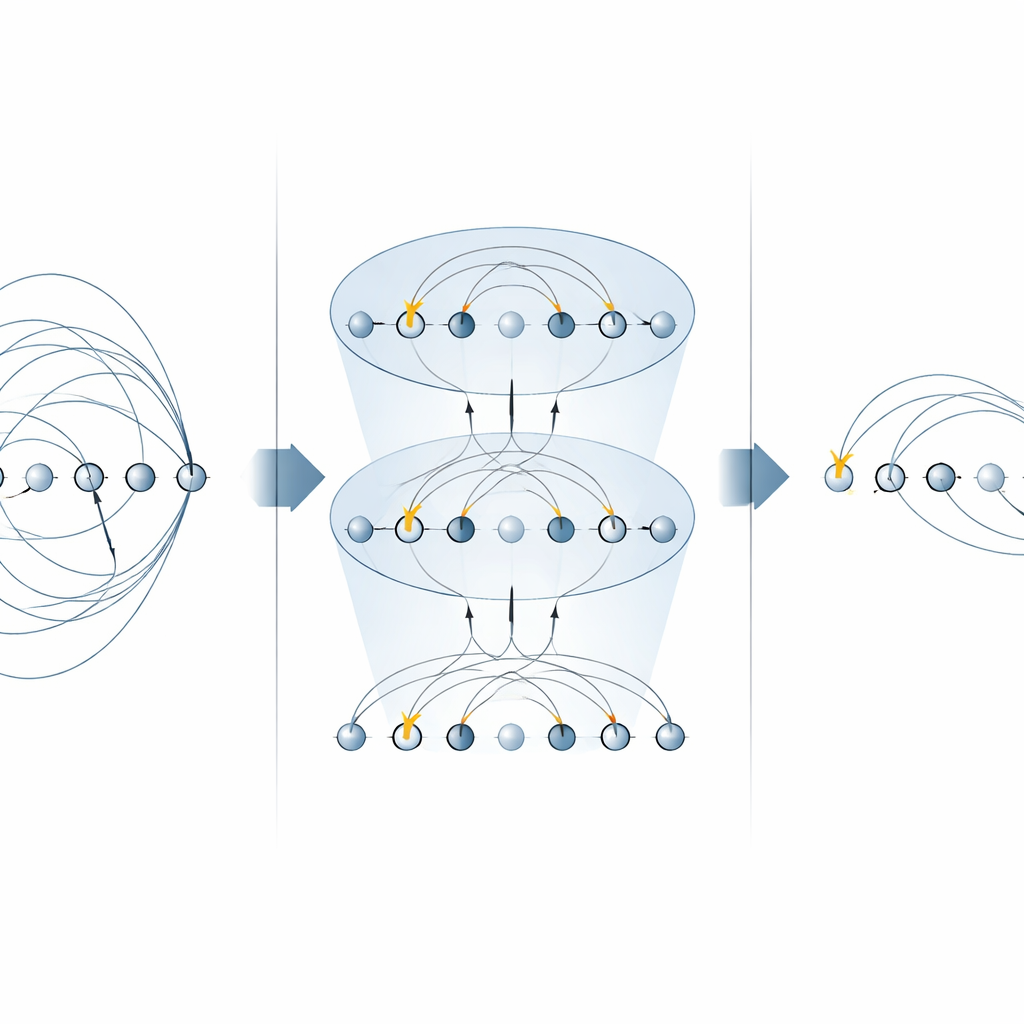
Att bygga snabbare kretsar för kvantkemi
Med dessa optimerade byggstenar konstruerar författarna kompletta kretsar både för variella grundtillståndssökningar och för verkliga tidsdynamiker. De illustrerar metoden på en liten men icke‑trivial molekyljon, H3+, och visar hur man sätter ihop ett helt UCCSD‑lager (singlar och dubblar) och ett Trotter‑steg för tidsutveckling med avsevärt färre globala grindar än tidigare tillvägagångssätt. Samma strategi generaliserar till högre ordningens excitationer, ger fördelar för alternativa ”qubit‑excitation”‑ansatser som är populära för näromsorgens enheter, och kan återanvändas för att direkt simulera elektroniska Hamiltonianer. Viktigt är att tillvägagångssättet respekterar centrala fysiska kvantiteter såsom partikelnumer och spinn, vilka är avgörande för kemiska tillämpningar.
Testa prestanda under realistiskt brus
Kortare kretsar hjälper bara om de faktiskt minskar felen på verklig hårdvara. För att kontrollera detta byggde teamet en detaljerad brusmodell av en 12‑jons linjär fångst, inklusive fluktuationer i vibrationsmodsfrekvenser och laserenergier—stora felkällor i dagens experiment. De jämförde sedan sina nya kretsar med standardkretsar för en rad små molekyler, och följde energifel, förlust av kvantfidelitet och överträdelser av bevarade storheter. För både singel‑ och dubbelexcitationer minskade de förbättrade designerna kretsinfideliteten med ungefär en halv till en hel storleksordning. För fullständiga molekylära beräkningar förde de konsekvent simulerade energier och fysiska observerbara närmare ideala resultat, och deras fördel blev tydligare för mer komplexa excitationer och större system.
Vad detta betyder för framtida simuleringar
Studien påstår inte att perfekt kvantkemi ligger runt hörnet; med dagens brusnivåer når även de förbättrade kretsarna ofta inte upp till att ensamma överträffa de bästa klassiska approximationerna. Men arbetet visar att genom att anpassa algoritmer till hårdvara—i detta fall genom att matcha fermionisk excitationsstruktur med globala jonfångstinteraktioner—kan man dramatiskt minska overhead och förbättra noggrannheten. Kombinerat med felbegränsningstekniker och mer måttfulla approximationer såsom qubit‑baserade excitationer, kan dessa strategier göra det möjligt för nästliga fångade‑jon‑enheter att lösa kemiskt relevanta problem som ligger precis bortom räckhåll för klassiska datorer.
Citering: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Nyckelord: fångade joners kvantdatorer, fermionisk simulering, molekylär kvantkemi, Mølmer–Sørensen‑grindar, unitär kopplad kluster