Clear Sky Science · en
Improved strategies for fermionic quantum simulation with global interactions
Why speeding up quantum chemistry matters
Designing new medicines, better batteries, and advanced materials often comes down to understanding how electrons move and interact inside molecules. Classical supercomputers quickly run out of steam when they try to track these many‑electron systems exactly. Trapped‑ion quantum computers promise a way forward, but today their calculations are still slow and noisy. This paper shows how to use the natural strengths of trapped ions to run a key class of chemistry calculations with far fewer operations, bringing accurate quantum simulations a step closer to practical reality.
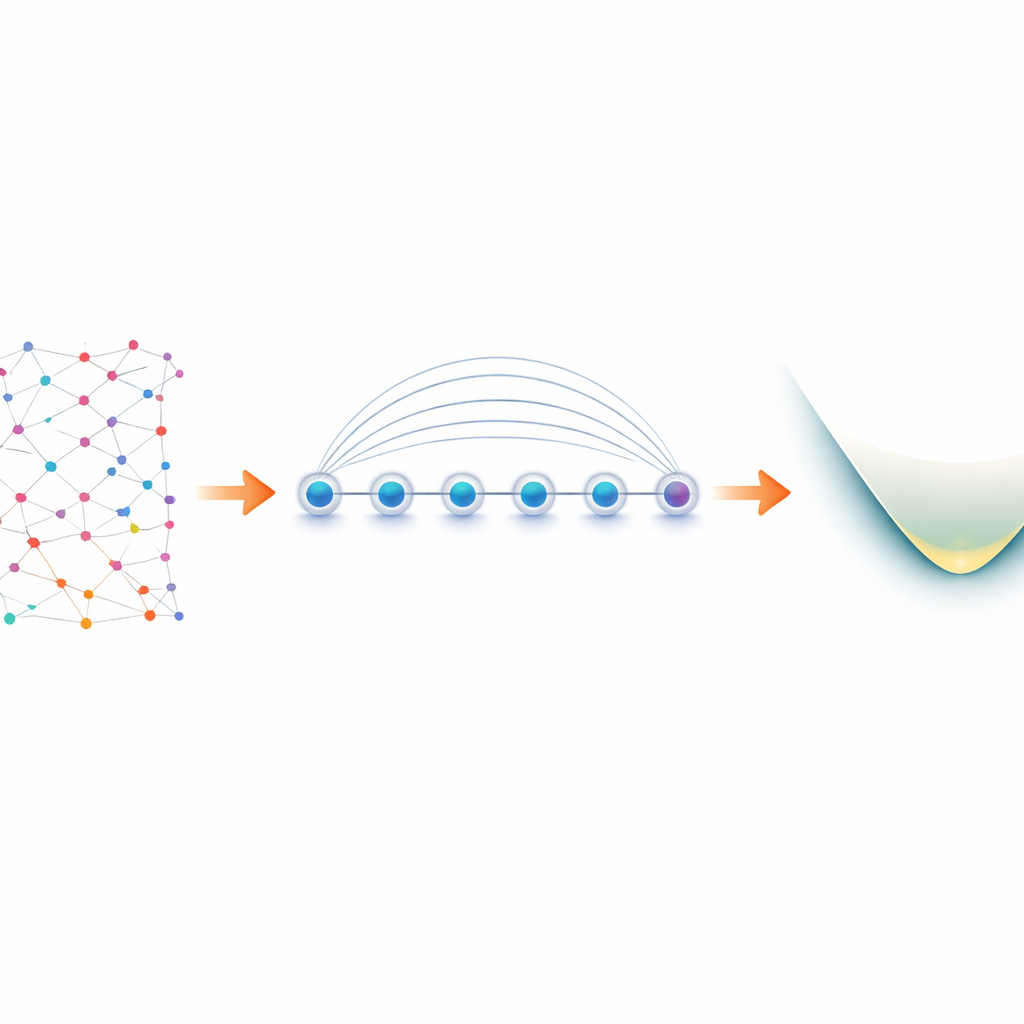
From electrons in molecules to qubits in the lab
Electrons in a molecule behave as “fermions,” following strict rules about how they can share quantum states. To simulate them on a quantum computer, researchers first translate these fermionic rules into operations on qubits, a process known as mapping. A popular choice, the Jordan–Wigner mapping, is conceptually simple but produces long‑range interactions: one logical operation on electrons can become a string of coupled qubits stretched across the entire device. On most quantum hardware, which only allows neighboring qubits to talk directly, this leads to circuit bloat and extra error‑prone swap operations. Trapped‑ion devices are different. Ions arranged in a line can be entangled all at once using a native operation called the Mølmer–Sørensen (MS) gate, which naturally connects distant qubits. The authors exploit this global interaction to turn Jordan–Wigner’s apparent weakness into a strength.
Using global interactions as a shortcut
The heart of many chemistry algorithms is the “excitation operator,” which describes moving electrons from occupied to empty orbitals. These operators appear in two major places: the unitary coupled‑cluster (UCC) method for finding molecular ground states, and in step‑by‑step (Trotterized) simulations of how an electronic system evolves in time. Previous schemes on trapped‑ion machines implemented each piece of an excitation operator separately, using multiple MS gates for every term. In this work, the authors show that specific forms of the MS gate can diagonalize whole families of these pieces at once. By placing simple single‑qubit rotations between just two MS gates, they are able to apply many non‑local components in parallel. For single‑electron excitations this cuts the number of required MS gates in half, and for double‑electron excitations it reduces them by a factor of four, without needing any extra helper qubits.
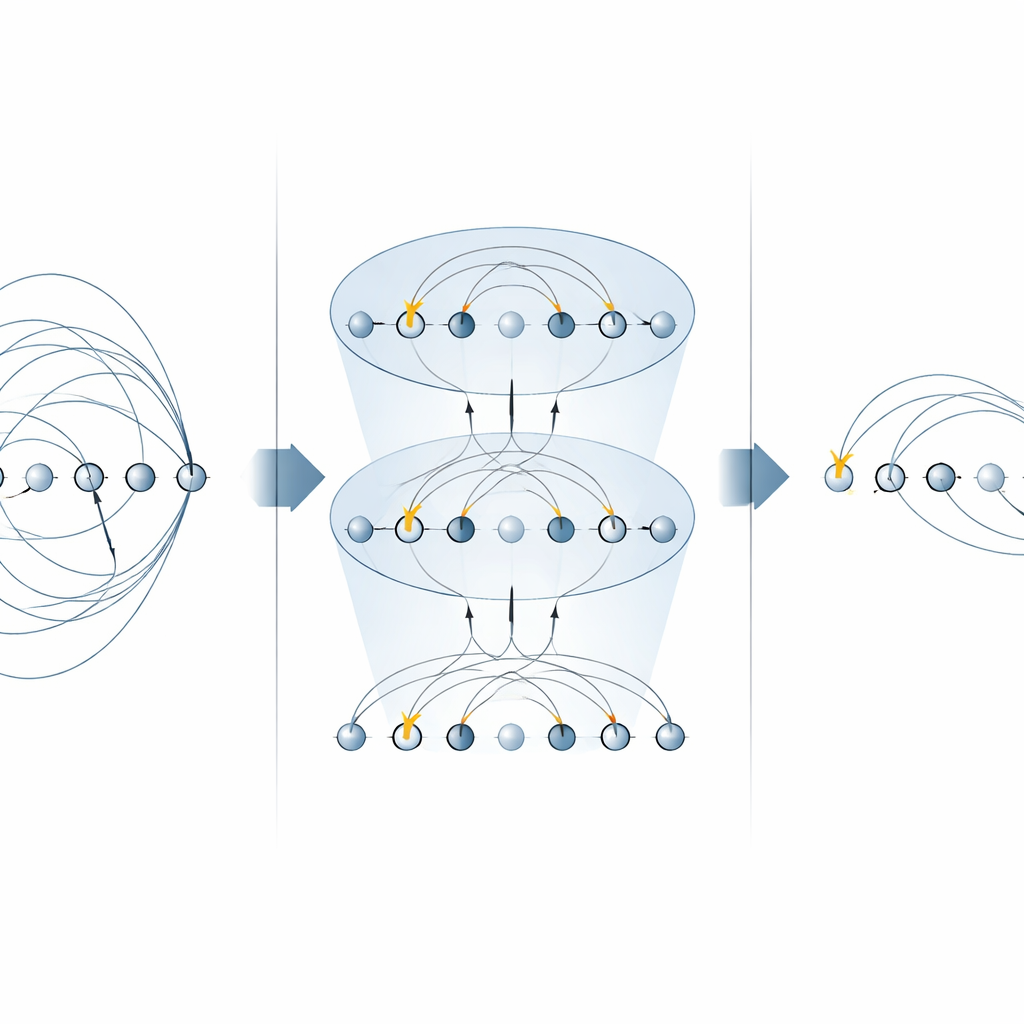
Building faster quantum chemistry circuits
With these optimized building blocks, the authors construct complete circuits for both variational ground‑state searches and real‑time dynamics. They illustrate the method on a small but non‑trivial molecular ion, H3+, showing how to assemble an entire UCCSD (singles‑and‑doubles) layer and a Trotter step for time evolution using far fewer global gates than earlier approaches. The same strategy generalizes to higher‑order excitations, brings benefits to alternative “qubit excitation” ansätze that are popular for near‑term devices, and can be reused for simulating electronic Hamiltonians directly. Importantly, the approach respects key physical quantities such as particle number and spin, which are central to chemistry applications.
Testing performance under realistic noise
Shorter circuits only help if they actually reduce errors on real hardware. To check this, the team built a detailed noise model of a 12‑ion linear trap, including fluctuations in vibrational mode frequencies and laser powers—major error sources in today’s experiments. They then compared their new circuits with standard ones for a range of small molecules, tracking energy errors, loss of quantum fidelity, and violations of conserved quantities. Across single and double excitations, the improved designs reduced circuit infidelity by roughly half to a full order of magnitude. For full molecular calculations, they consistently brought simulated energies and physical observables closer to ideal results, and their advantage grew more pronounced for more complex excitations and larger systems.
What this means for future simulations
The study does not claim that perfect quantum chemistry is around the corner; with current noise levels, even the improved circuits often fall short of surpassing the best classical approximations by themselves. However, the work demonstrates that by matching algorithms to hardware—in this case, aligning fermionic excitation structure with global ion‑trap interactions—one can dramatically reduce overhead and improve accuracy. Combined with error‑mitigation techniques and more modest approximations such as qubit‑based excitations, these strategies could enable near‑term trapped‑ion devices to tackle chemically relevant problems that are just beyond the reach of classical computers.
Citation: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Keywords: trapped ion quantum computing, fermionic simulation, molecular quantum chemistry, Mølmer–Sørensen gates, unitary coupled cluster