Clear Sky Science · pt
Estratégias aprimoradas para simulação quântica fermiónica com interações globais
Por que acelerar a química quântica importa
Projetar novos medicamentos, baterias melhores e materiais avançados muitas vezes depende de entender como os elétrons se movem e interagem dentro de moléculas. Supercomputadores clássicos rapidamente ficam sem fôlego quando tentam acompanhar esses sistemas com muitos elétrons exatamente. Computadores quânticos com íons aprisionados prometem um caminho adiante, mas hoje seus cálculos ainda são lentos e ruidosos. Este artigo mostra como usar as forças naturais dos íons aprisionados para executar uma classe chave de cálculos de química com muito menos operações, aproximando simulações quânticas precisas de uma realidade prática.
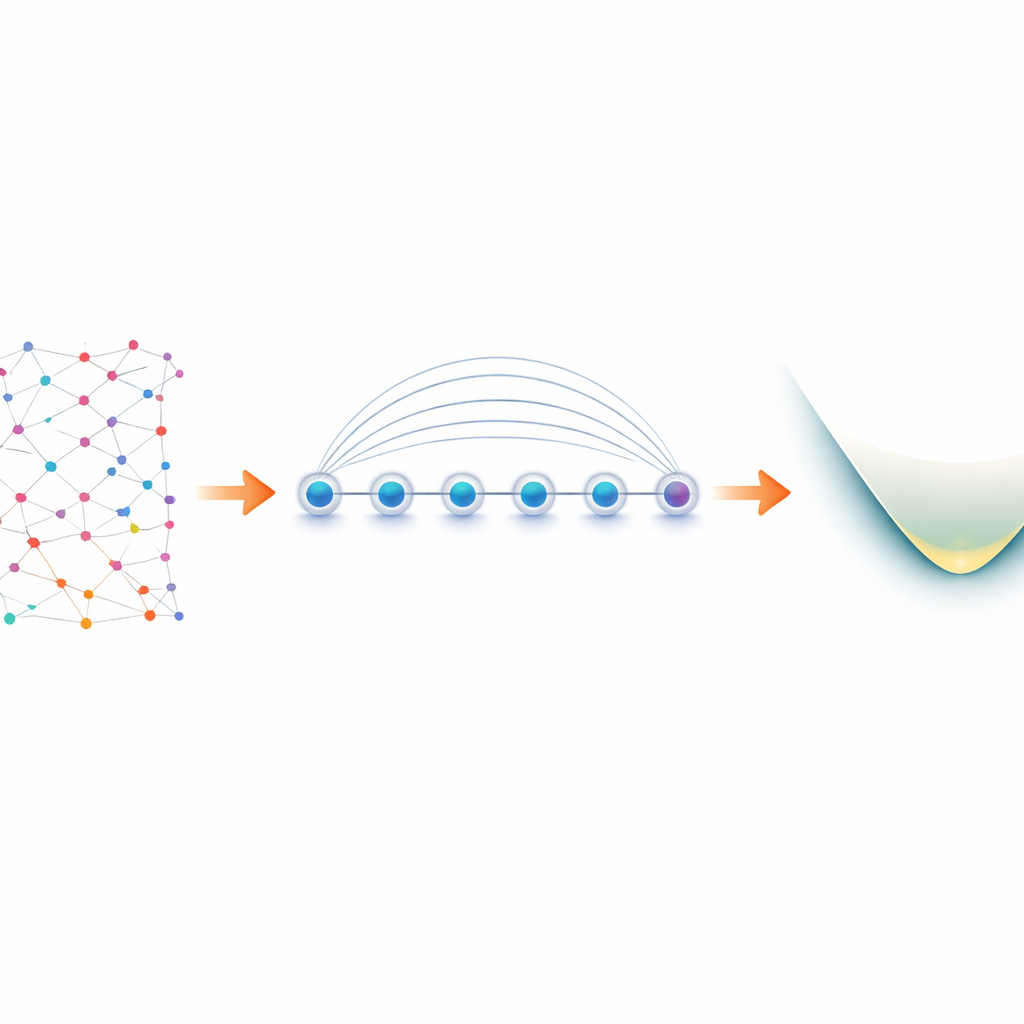
Dos elétrons nas moléculas aos qubits no laboratório
Elétrons em uma molécula se comportam como “férmions”, seguindo regras rígidas sobre como podem compartilhar estados quânticos. Para simulá‑los em um computador quântico, os pesquisadores primeiro traduzem essas regras fermiónicas em operações sobre qubits, um processo conhecido como mapeamento. Uma escolha popular, o mapeamento de Jordan–Wigner, é conceitualmente simples, mas gera interações de longo alcance: uma operação lógica nos elétrons pode se tornar uma cadeia de qubits acoplados estendida através de todo o dispositivo. Na maioria do hardware quântico, que só permite que qubits vizinhos se comuniquem diretamente, isso leva a inchaço de circuito e operações de swap extras sujeitas a erros. Dispositivos de íons aprisionados são diferentes. Íons dispostos em linha podem ser emaranhados de uma vez só usando uma operação nativa chamada porta Mølmer–Sørensen (MS), que conecta naturalmente qubits distantes. Os autores exploram essa interação global para transformar a aparente fraqueza de Jordan–Wigner em uma vantagem.
Usando interações globais como atalho
O cerne de muitos algoritmos de química é o “operador de excitação”, que descreve mover elétrons de orbitais ocupados para vazios. Esses operadores aparecem em dois lugares principais: o método de acoplamento unitário por cluster (UCC) para encontrar estados fundamentais moleculares, e em simulações passo a passo (trotterizadas) de como um sistema eletrônico evolui no tempo. Esquemas anteriores em máquinas de íons aprisionados implementavam cada peça de um operador de excitação separadamente, usando múltiplas portas MS para cada termo. Neste trabalho, os autores mostram que formas específicas da porta MS podem diagonalizar famílias inteiras desses termos de uma só vez. Ao colocar rotações simples de qubit único entre apenas duas portas MS, eles conseguem aplicar muitos componentes não locais em paralelo. Para excitações de elétron único isso corta o número de portas MS necessárias pela metade, e para excitações de elétron duplo reduz por um fator de quatro, sem precisar de qubits auxiliares extras.
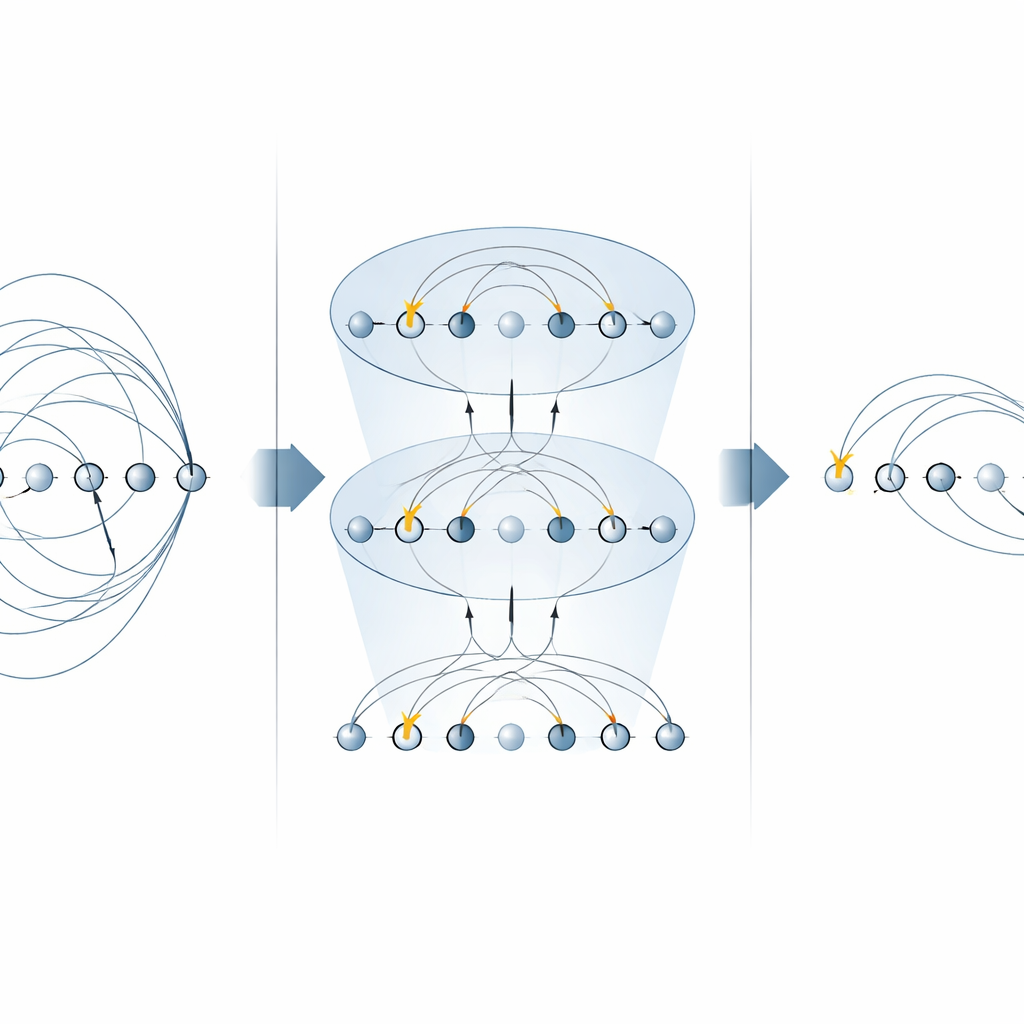
Construindo circuitos de química quântica mais rápidos
Com esses blocos de construção otimizados, os autores constroem circuitos completos tanto para buscas variacionais do estado fundamental quanto para dinâmica em tempo real. Eles ilustram o método em um íon molecular pequeno mas não trivial, H3+, mostrando como montar uma camada inteira UCCSD (singulares e duplos) e um passo de Trotter para evolução temporal usando muito menos portas globais do que abordagens anteriores. A mesma estratégia generaliza para excitações de ordem superior, traz benefícios a anstazes alternativas de “excitação de qubit” que são populares para dispositivos de curto prazo, e pode ser reutilizada para simular hamiltonianos eletrônicos diretamente. Importante, a abordagem respeita quantidades físicas chave como o número de partículas e o spin, que são centrais para aplicações em química.
Testando desempenho sob ruído realista
Circuitos mais curtos só ajudam se realmente reduzirem erros no hardware real. Para verificar isso, a equipe construiu um modelo de ruído detalhado de uma armadilha linear de 12 íons, incluindo flutuações nas frequências dos modos vibracionais e nas potências dos lasers — fontes principais de erro nos experimentos atuais. Eles então compararam seus novos circuitos com os padrão para uma gama de moléculas pequenas, acompanhando erros de energia, perda de fidelidade quântica e violações de quantidades conservadas. Em excitações simples e duplas, os projetos melhorados reduziram a infidelidade dos circuitos em aproximadamente metade a uma ordem de magnitude completa. Para cálculos moleculares completos, eles consistentemente trouxeram energias simuladas e observáveis físicos mais próximos dos resultados ideais, e sua vantagem se tornou mais pronunciada para excitações mais complexas e sistemas maiores.
O que isso significa para simulações futuras
O estudo não afirma que a química quântica perfeita esteja ao virar da esquina; com os níveis de ruído atuais, mesmo os circuitos aprimorados muitas vezes ficam aquém de superar, por si só, as melhores aproximações clássicas. No entanto, o trabalho demonstra que, ao alinhar algoritmos ao hardware — neste caso, ajustando a estrutura de excitação fermiónica às interações globais da armadilha de íons — é possível reduzir drasticamente a sobrecarga e melhorar a precisão. Combinadas com técnicas de mitigação de erro e aproximações mais modestas, como excitações baseadas em qubits, essas estratégias poderiam permitir que dispositivos de íons aprisionados de curto prazo enfrentem problemas quimicamente relevantes que estão além do alcance dos computadores clássicos.
Citação: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Palavras-chave: computação quântica com íons aprisionados, simulação fermiónica, química quântica molecular, portas Mølmer–Sørensen, acoplamento unitário por cluster