Clear Sky Science · es
Estrategias mejoradas para la simulación cuántica fermiónica con interacciones globales
Por qué acelerar la química cuántica es importante
Diseñar nuevos medicamentos, baterías más eficientes y materiales avanzados suele reducirse a comprender cómo se mueven e interactúan los electrones dentro de las moléculas. Las supercomputadoras clásicas se quedan pronto sin recursos cuando intentan seguir con exactitud estos sistemas con muchos electrones. Los ordenadores cuánticos de iones atrapados prometen una vía de avance, pero hoy sus cálculos siguen siendo lentos y ruidosos. Este trabajo muestra cómo aprovechar las fortalezas naturales de los iones atrapados para ejecutar una clase clave de cálculos de química con muchas menos operaciones, acercando las simulaciones cuánticas precisas a una realidad práctica.
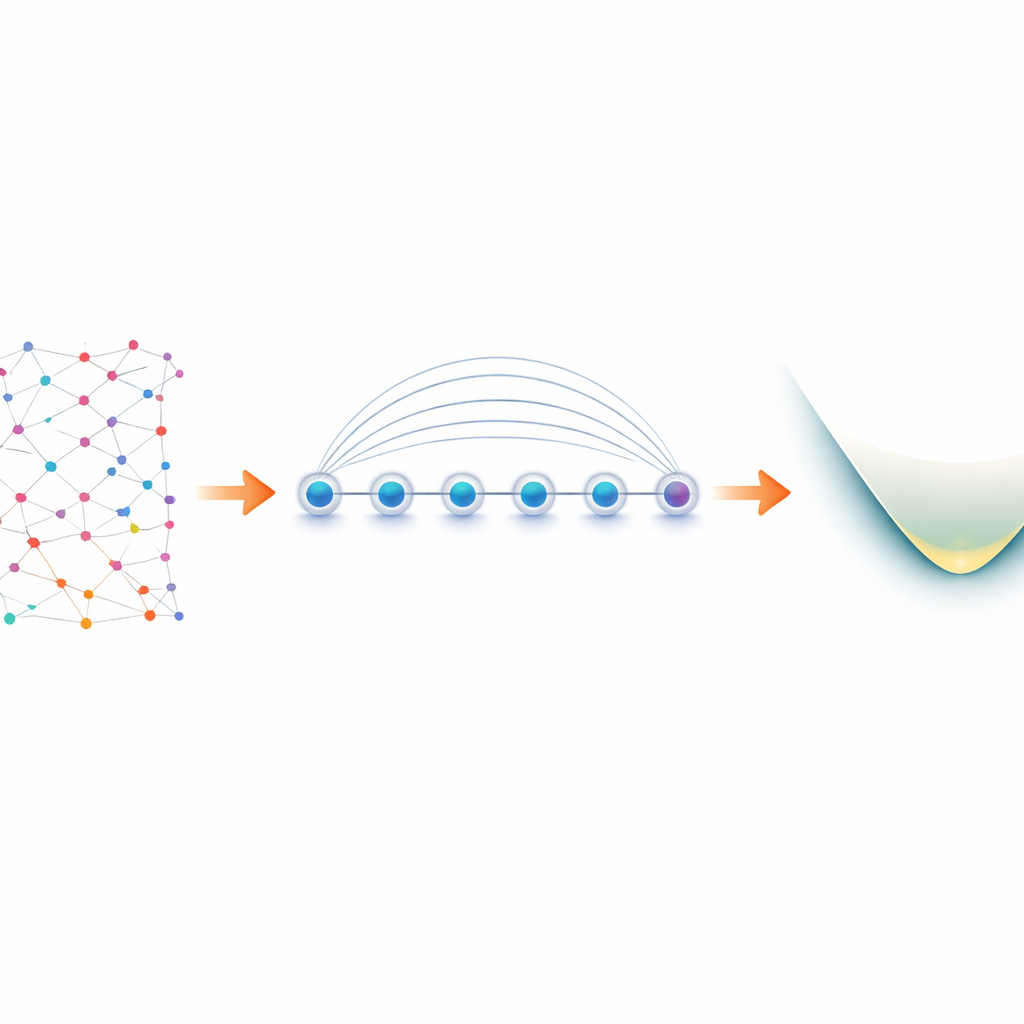
De los electrones en moléculas a los qubits en el laboratorio
Los electrones en una molécula se comportan como “fermiones”, siguiendo reglas estrictas sobre cómo pueden compartir estados cuánticos. Para simulárlos en un ordenador cuántico, los investigadores primero traducen estas reglas fermiónicas a operaciones sobre qubits, un proceso conocido como mapeo. Una elección popular, el mapeo de Jordan–Wigner, es conceptualmente simple pero produce interacciones de largo alcance: una operación lógica sobre electrones puede convertirse en una cadena de qubits acoplados que se extiende a lo largo del dispositivo. En la mayoría del hardware cuántico, que solo permite comunicación directa entre qubits vecinos, esto conduce a un ensanchamiento del circuito y a operaciones de intercambio (swap) adicionales, propensas a errores. Los dispositivos de iones atrapados son diferentes. Iones dispuestos en línea pueden entrelazarse a la vez mediante una operación nativa llamada puerta Mølmer–Sørensen (MS), que conecta de forma natural qubits distantes. Los autores explotan esta interacción global para convertir la aparente debilidad de Jordan–Wigner en una ventaja.
Usar interacciones globales como atajo
El núcleo de muchos algoritmos de química es el «operador de excitación», que describe el movimiento de electrones desde orbitales ocupados a orbitales vacíos. Estos operadores aparecen en dos lugares principales: el método de acoplamiento unitario por clústeres (UCC) para encontrar estados fundamentales moleculares, y en simulaciones paso a paso (trotterizadas) de cómo evoluciona un sistema electrónico en el tiempo. Los esquemas previos en máquinas de iones atrapados implementaban cada pieza de un operador de excitación por separado, usando múltiples puertas MS para cada término. En este trabajo, los autores muestran que formas específicas de la puerta MS pueden diagonalizar familias enteras de estos términos de una sola vez. Colocando rotaciones sencillas de un solo qubit entre solo dos puertas MS, consiguen aplicar muchos componentes no locales en paralelo. Para excitaciones de un solo electrón, esto reduce a la mitad el número de puertas MS necesarias, y para excitaciones de doble electrón lo reduce por un factor de cuatro, sin necesitar qubits auxiliares adicionales.
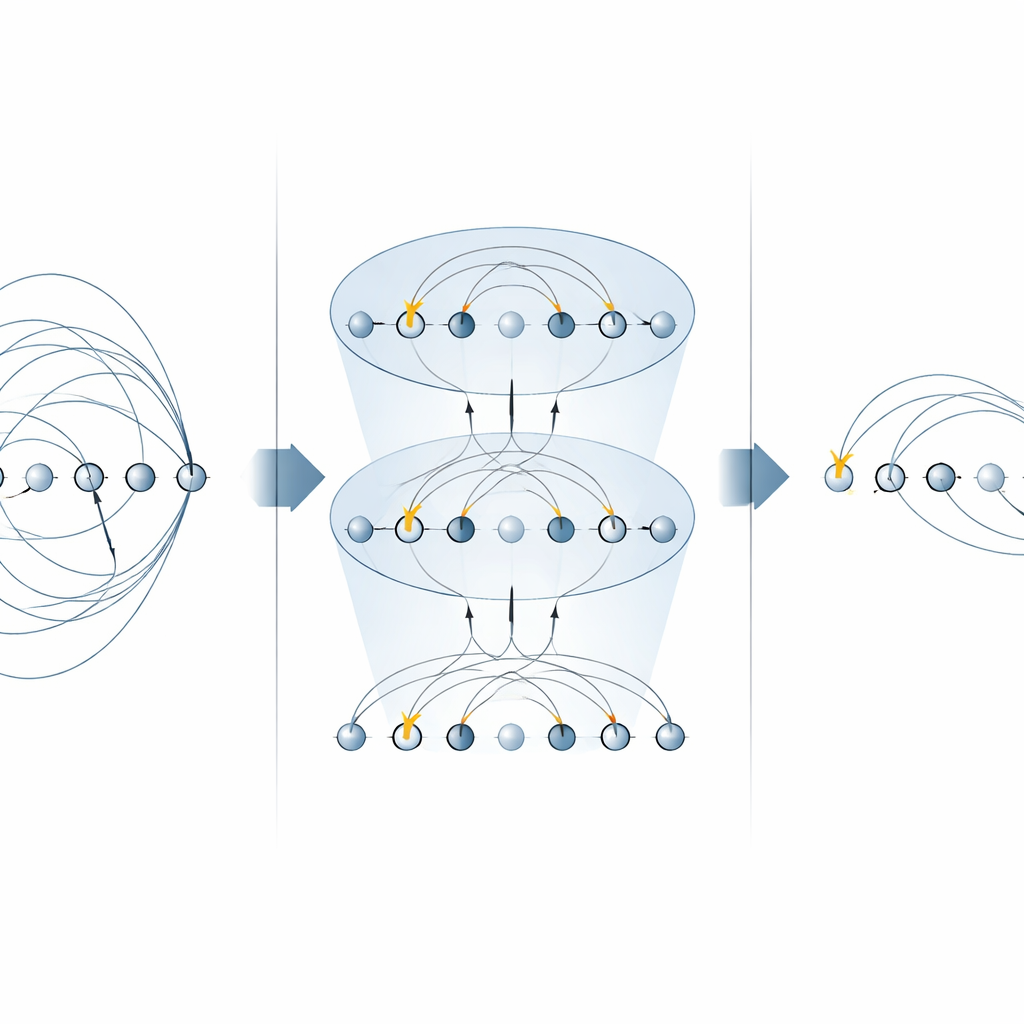
Construir circuitos de química cuántica más rápidos
Con estos bloques construidos optimizados, los autores ensamblan circuitos completos tanto para búsquedas variacionales del estado fundamental como para dinámica en tiempo real. Ilustran el método en un ión molecular pequeño pero no trivial, H3+, mostrando cómo montar una capa UCCSD (exciciones singulares y dobles) completa y un paso de Trotter para la evolución temporal usando muchas menos puertas globales que enfoques anteriores. La misma estrategia se generaliza a excitaciones de orden superior, beneficia a ansätze alternativos de «excitación de qubits» populares en dispositivos de corto plazo, y puede reutilizarse para simular directamente hamiltonianos electrónicos. Es importante que el enfoque respeta cantidades físicas clave como el número de partículas y el espín, centrales en aplicaciones químicas.
Probar el rendimiento bajo ruido realista
Los circuitos más cortos solo ayudan si realmente reducen los errores en hardware real. Para comprobarlo, el equipo construyó un modelo detallado de ruido de una trampa lineal de 12 iones, incluyendo fluctuaciones en las frecuencias de modos vibracionales y en las potencias láser, fuentes de error importantes en los experimentos actuales. Luego compararon sus nuevos circuitos con los estándar para una gama de moléculas pequeñas, siguiendo los errores de energía, la pérdida de fidelidad cuántica y las violaciones de cantidades conservadas. Tanto en excitaciones simples como dobles, los diseños mejorados redujeron la infidelidad del circuito aproximadamente a la mitad hasta un orden de magnitud completo. Para cálculos moleculares completos, consistentemente acercaron las energías simuladas y los observables físicos a los resultados ideales, y su ventaja se hizo más pronunciada para excitaciones más complejas y sistemas mayores.
Qué significa esto para futuras simulaciones
El estudio no afirma que la química cuántica perfecta esté a la vuelta de la esquina; con los niveles de ruido actuales, incluso los circuitos mejorados a menudo no alcanzan por sí solos a superar las mejores aproximaciones clásicas. Sin embargo, el trabajo demuestra que al ajustar los algoritmos al hardware—en este caso, alineando la estructura de las excitaciones fermiónicas con las interacciones globales de las trampas de iones—se puede reducir drásticamente la sobrecarga y mejorar la precisión. Combinadas con técnicas de mitigación de errores y aproximaciones más moderadas, como las excitaciones basadas en qubits, estas estrategias podrían permitir a dispositivos de iones atrapados de corto plazo abordar problemas químicamente relevantes que están justo más allá del alcance de los ordenadores clásicos.
Cita: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Palabras clave: computación cuántica con iones atrapados, simulación fermiónica, química cuántica molecular, puertas Mølmer–Sørensen, acople unitario por clústeres