Clear Sky Science · fr
Stratégies améliorées pour la simulation quantique fermionique avec interactions globales
Pourquoi accélérer la chimie quantique est important
Concevoir de nouveaux médicaments, de meilleures batteries ou des matériaux avancés revient souvent à comprendre comment les électrons se déplacent et interagissent au sein des molécules. Les supercalculateurs classiques s’épuisent rapidement lorsqu’il s’agit de suivre exactement ces systèmes à nombreux électrons. Les ordinateurs quantiques à ions piégés offrent une voie prometteuse, mais aujourd’hui leurs calculs restent lents et bruyants. Cet article montre comment exploiter les forces naturelles des ions piégés pour exécuter une classe clé de calculs de chimie avec beaucoup moins d’opérations, rapprochant ainsi les simulations quantiques précises d’une réalité pratique.
Des électrons dans les molécules aux qubits en laboratoire
Les électrons dans une molécule se comportent comme des « fermions », suivant des règles strictes sur la façon dont ils peuvent partager des états quantiques. Pour les simuler sur un ordinateur quantique, les chercheurs traduisent d’abord ces règles fermioniques en opérations sur des qubits, un processus appelé mappage. Un choix populaire, le mappage de Jordan–Wigner, est conceptuellement simple mais produit des interactions de grande portée : une opération logique sur les électrons peut devenir une chaîne de qubits couplés s’étirant sur tout l’appareil. Sur la plupart des matériels quantiques, qui n’autorisent que des interactions directes entre qubits voisins, cela entraîne un gonflement des circuits et des opérations d’échange (swap) supplémentaires, sources d’erreurs. Les dispositifs à ions piégés sont différents. Des ions alignés peuvent être intriqués simultanément à l’aide d’une opération native appelée porte de Mølmer–Sørensen (MS), qui relie naturellement des qubits distants. Les auteurs exploitent cette interaction globale pour transformer la faiblesse apparente de Jordan–Wigner en un avantage.
Utiliser les interactions globales comme raccourci
Le cœur de nombreux algorithmes de chimie est l’« opérateur d’excitation », qui décrit le déplacement d’électrons d’orbitales occupées vers des orbitales vides. Ces opérateurs apparaissent à deux endroits principaux : la méthode unitary coupled‑cluster (UCC) pour trouver les états fondamentaux moléculaires, et dans les simulations pas à pas (trotterisées) de l’évolution temporelle d’un système électronique. Les schémas précédents sur machines à ions piégés implémentaient chaque composante d’un opérateur d’excitation séparément, en utilisant plusieurs portes MS pour chaque terme. Dans ce travail, les auteurs montrent que des formes spécifiques de la porte MS peuvent diagonaliser des familles entières de ces composantes en une seule fois. En plaçant de simples rotations sur un seul qubit entre seulement deux portes MS, ils peuvent appliquer en parallèle de nombreux composants non locaux. Pour les excitations monoélectroniques, cela divise par deux le nombre de portes MS nécessaires, et pour les excitations diélectroniques, cela le réduit d’un facteur quatre, sans nécessité de qubits auxiliaires supplémentaires.
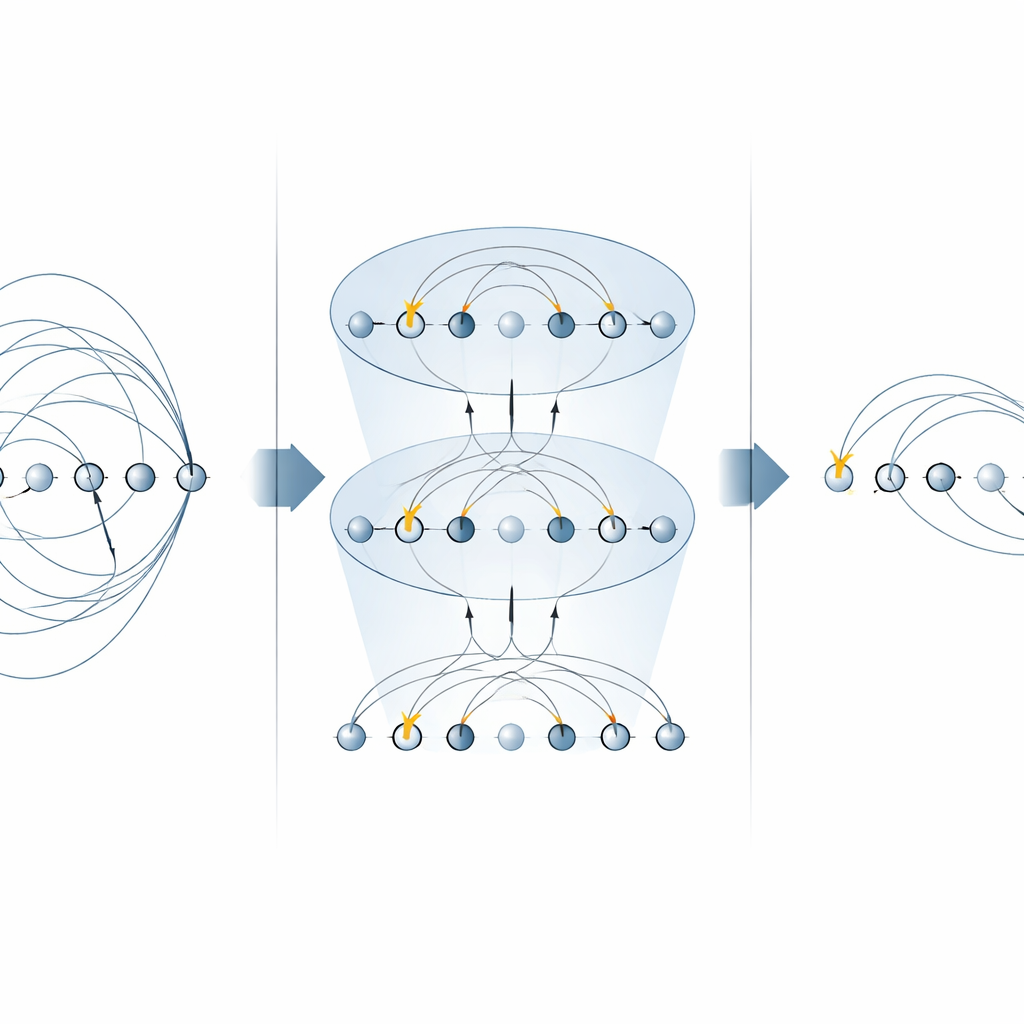
Construire des circuits de chimie quantique plus rapides
Avec ces blocs de construction optimisés, les auteurs construisent des circuits complets pour la recherche variationnelle d’états fondamentaux et pour la dynamique en temps réel. Ils illustrent la méthode sur un ion moléculaire petit mais non trivial, H3+, montrant comment assembler une couche UCCSD entière (singles‑and‑doubles) et un pas de Trotter pour l’évolution temporelle en utilisant bien moins de portes globales que les approches antérieures. La même stratégie se généralise aux excitations d’ordre supérieur, apporte des bénéfices aux ansatz alternatifs de « qubit excitation » populaires pour les dispositifs à court terme, et peut être réutilisée pour simuler directement des hamiltoniens électroniques. Fait important, l’approche respecte des grandeurs physiques clés telles que le nombre de particules et le spin, qui sont centrales pour les applications en chimie.
Tester les performances en présence de bruit réaliste
Des circuits plus courts n’aident que s’ils réduisent effectivement les erreurs sur du matériel réel. Pour vérifier cela, l’équipe a construit un modèle de bruit détaillé d’un piège linéaire de 12 ions, incluant les fluctuations des fréquences des modes vibrationnels et des puissances laser — principales sources d’erreurs dans les expériences actuelles. Ils ont ensuite comparé leurs nouveaux circuits aux circuits standard pour une série de petites molécules, en suivant les erreurs d’énergie, la perte de fidélité quantique et les violations des quantités conservées. Pour les excitations simples et doubles, les conceptions améliorées ont réduit l’infidélité des circuits d’environ moitié jusqu’à un ordre de grandeur. Pour des calculs moléculaires complets, elles ont systématiquement rapproché les énergies simulées et les observables physiques des résultats idéaux, et leur avantage s’est avéré plus marqué pour des excitations plus complexes et des systèmes de plus grande taille.
Ce que cela signifie pour les simulations futures
L’étude ne prétend pas que la chimie quantique parfaite soit imminente ; avec les niveaux de bruit actuels, même les circuits améliorés n’arrivent souvent pas à surpasser à eux seuls les meilleures approximations classiques. Cependant, le travail montre qu’en adaptant les algorithmes au matériel — dans ce cas en alignant la structure des excitations fermioniques avec les interactions globales des pièges d’ions — on peut réduire drastiquement les surcoûts et améliorer la précision. Associées à des techniques d’atténuation d’erreurs et à des approximations plus modestes comme les excitations basées sur les qubits, ces stratégies pourraient permettre aux dispositifs à ions piégés à court terme d’aborder des problèmes chimiquement pertinents qui dépassent actuellement la portée des ordinateurs classiques.
Citation: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Mots-clés: informatique quantique sur ions piégés, simulation fermionique, chimie quantique moléculaire, portes de Mølmer–Sørensen, couplage unitaire en cluster