Clear Sky Science · ru
Улучшенные стратегии для фермионной квантовой симуляции с глобальными взаимодействиями
Почему ускорение квантовой химии важно
Проектирование новых лекарств, более совершенных аккумуляторов и продвинутых материалов во многом сводится к пониманию того, как электроны движутся и взаимодействуют внутри молекул. Классические Суперкомпьютеры быстро исчерпывают свои возможности при попытке точно отслеживать такие многoэлектронные системы. Квантовые компьютеры на захваченных ионах предлагают путь вперёд, но сегодня их расчёты всё ещё медленные и шумные. В этой статье показано, как использовать природные сильные стороны устройств на ионах для выполнения ключевого класса химических расчётов с гораздо меньшим числом операций, что приближает точные квантовые симуляции к практической реальности.
От электронов в молекулах к кубитам в лаборатории
Электроны в молекуле ведут себя как «фермионы», подчиняясь строгим правилам совместного занятия квантовых состояний. Чтобы моделировать их на квантовом компьютере, исследователи сначала переводят эти фермионные правила в операции над кубитами — процесс, известный как отображение. Популярный выбор, отображение Джордана–Вигнера, концептуально прост, но создаёт дальнодействующие взаимодействия: одна логическая операция для электронов может превратиться в цепочку связанных кубитов, растянутую по всему устройству. На большинстве квантовых аппаратных платформ, которые допускают прямое взаимодействие только соседних кубитов, это приводит к раздутым схемам и дополнительным ошибкоёмким операциям перестановки. У устройств на захваченных ионах ситуация иная. Ионы, расположенные в линию, можно спутать одновременно с помощью нативной операции, называемой воротами Мёлмера–Сёрсена (MS), которые естественным образом связывают удалённые кубиты. Авторы используют это глобальное взаимодействие, чтобы превратить кажущееся слабое место отображения Джордана–Вигнера в преимущество.
Использование глобальных взаимодействий как короткого пути
Сердцем многих химических алгоритмов является «оператор возбуждения», описывающий перенос электронов из заполненных орбиталей в пустые. Эти операторы появляются в двух основных местах: в унитарном связанном кластерном (UCC) методе для поиска молекулярного основного состояния и в поэтапных (троттеризованных) симуляциях временной эволюции электронной системы. Предыдущие схемы на устройствах с захваченными ионами реализовывали каждую часть оператора возбуждения по отдельности, используя несколько MS-входов для каждого терма. В этой работе авторы показывают, что специальные формы MS-врат могут диагонализировать целые семейства таких слагаемых одновременно. Размещая простые однокубитные повороты между всего двумя MS-вратами, они могут применять многие нелокальные компоненты параллельно. Для однок-электронных возбуждений это сокращает число требуемых MS-врат вдвое, а для двухэлектронных — сокращает их в четыре раза, без необходимости дополнительных вспомогательных кубитов.
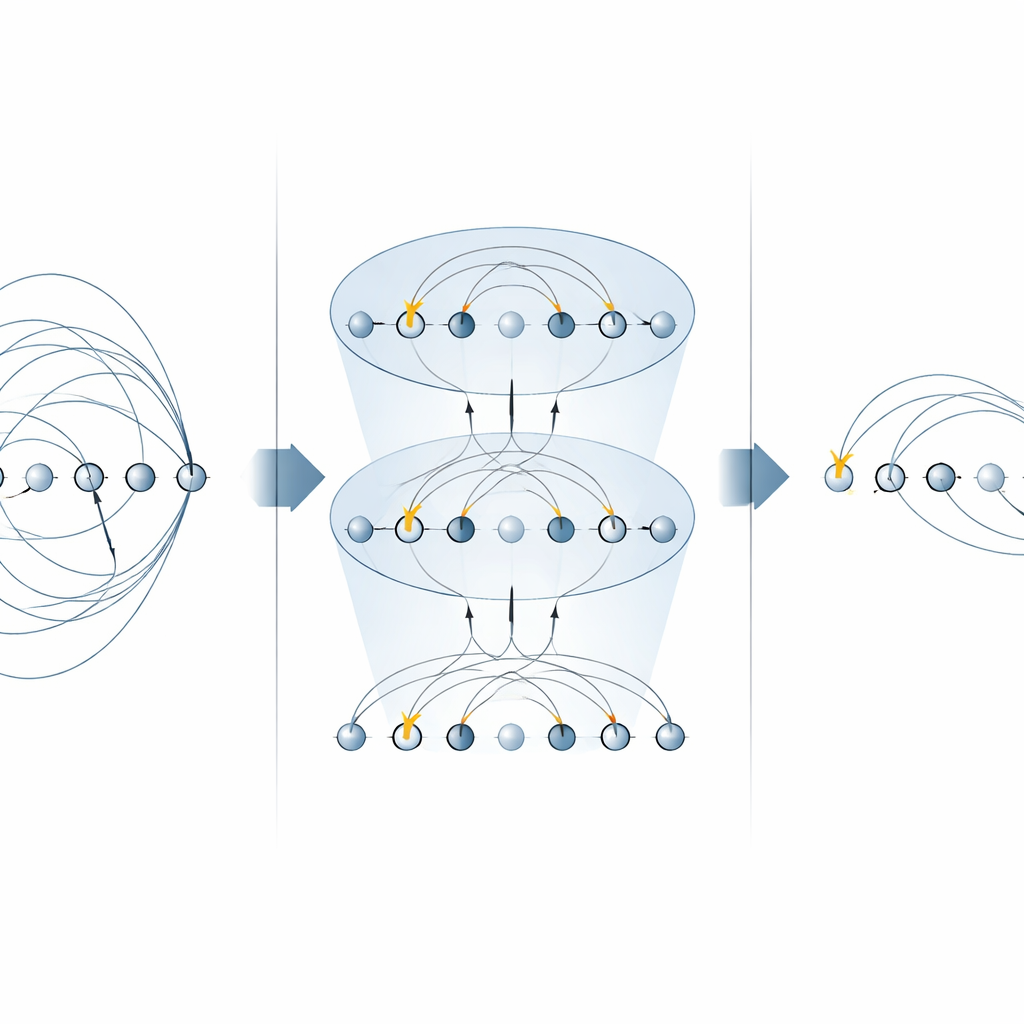
Построение более быстрых схем квантовой химии
Используя эти оптимизированные блоки, авторы конструируют полные схемы как для вариационного поиска основного состояния, так и для реального времени динамики. Они иллюстрируют метод на небольшом, но нетривиальном молекулярном ионе H3+, показывая, как собрать целый слой UCCSD (с учётом одиночных и парных возбуждений) и троттеровский шаг для эволюции во времени, используя значительно меньше глобальных ворот, чем в ранних подходах. Та же стратегия обобщается на возбуждения более высокого порядка, приносит выгоду альтернативным анзацам «возбуждения кубитов», популярным для устройств ближнего срока, и может быть повторно использована для прямой симуляции электронных гамильтонов. Важно, что подход сохраняет ключевые физические величины, такие как число частиц и спин, которые имеют центральное значение в химических приложениях.
Тестирование производительности при реалистичном шуме
Короткие схемы помогают лишь в том случае, если они действительно уменьшают ошибки на реальном аппаратном обеспечении. Чтобы проверить это, команда построила детальную модель шума для линейной ловушки из 12 ионов, включая флуктуации частот вибрационных мод и мощностей лазеров — основные источники ошибок в современных экспериментах. Затем они сравнили свои новые схемы со стандартными для ряда небольших молекул, отслеживая ошибки в энергии, потерю квантовой точности и нарушения сохраняемых величин. Для одиночных и парных возбуждений улучшённые конструкции снизили нечёткость схем примерно в 2 раза и вплоть до целого порядка величины. Для полных молекулярных расчётов они последовательно приближали смоделированные энергии и физические наблюдаемые к идеальным результатам, и их преимущество становилось более выраженным для более сложных возбуждений и больших систем.
Что это значит для будущих симуляций
Исследование не утверждает, что совершенная квантовая химия уже не за горами; при текущих уровнях шума даже улучшённые схемы часто не в состоянии сами по себе превзойти лучшие классические приближения. Тем не менее работа демонстрирует, что согласование алгоритмов с аппаратурой — в данном случае выравнивание структуры фермионных возбуждений с глобальными взаимодействиями ионной ловушки — позволяет существенно сократить накладные расходы и повысить точность. В сочетании с техниками уменьшения ошибок и более сдержанными приближениями, такими как возбуждения в представлении кубитов, эти стратегии могут позволить устройствам на захваченных ионах ближнего срока решать химически значимые задачи, которые находятся чуть за пределами возможностей классических компьютеров.
Цитирование: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Ключевые слова: квантовые вычисления на захваченных ионах, фермионная симуляция, молекулярная квантовая химия, ворота Мёлмера–Сёрсена, унитарная связанная кластерная методика