Clear Sky Science · it
Strategie migliorate per la simulazione quantistica di fermioni con interazioni globali
Perché accelerare la chimica quantistica è importante
Progettare nuovi farmaci, batterie migliori e materiali avanzati spesso si riduce a capire come gli elettroni si muovono e interagiscono all’interno delle molecole. I supercomputer classici si esauriscono rapidamente quando cercano di tracciare esattamente questi sistemi a molti elettroni. I computer quantistici a ioni intrappolati promettono una via d’uscita, ma oggi i loro calcoli restano lenti e rumorosi. Questo articolo mostra come sfruttare i punti di forza naturali degli ioni intrappolati per eseguire una classe chiave di calcoli chimici con molte meno operazioni, avvicinando le simulazioni quantistiche accurate a una realtà pratica.
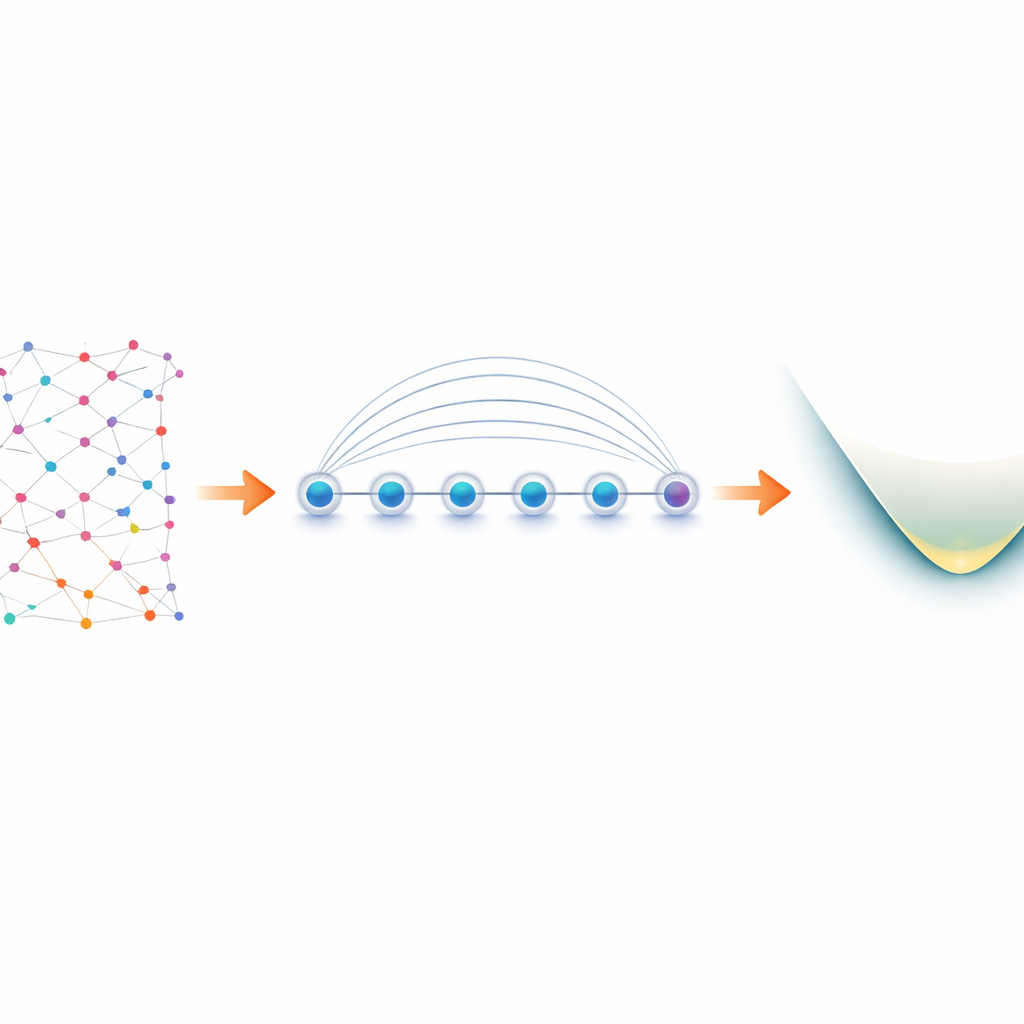
Dagli elettroni nelle molecole ai qubit in laboratorio
Gli elettroni in una molecola si comportano come “fermioni”, seguendo regole rigorose su come possono condividere gli stati quantistici. Per simularli su un computer quantistico, i ricercatori prima traducono queste regole fermioniche in operazioni sui qubit, un processo noto come mappatura. Una scelta popolare, la mappatura di Jordan–Wigner, è concettualmente semplice ma produce interazioni a lunga distanza: un’operazione logica sugli elettroni può diventare una stringa di qubit accoppiati estesa su tutto il dispositivo. Sulla maggior parte dell’hardware quantistico, che permette la comunicazione diretta solo tra qubit vicini, questo porta a circuiti gonfiati e a operazioni di swap aggiuntive soggette a errori. I dispositivi a ioni intrappolati sono diversi. Ioni disposti in linea possono essere entangled tutti insieme usando un’operazione nativa chiamata porta Mølmer–Sørensen (MS), che collega naturalmente qubit distanti. Gli autori sfruttano questa interazione globale per trasformare quella che sembra una debolezza della mappatura Jordan–Wigner in un punto di forza.
Usare le interazioni globali come scorciatoia
Il nucleo di molti algoritmi chimici è l’«operatore di eccitazione», che descrive lo spostamento di elettroni da orbitali occupati a orbitali vuoti. Questi operatori compaiono in due contesti principali: il metodo unitary coupled‑cluster (UCC) per trovare gli stati fondamentali molecolari, e nelle simulazioni passo per passo (trotterizzate) dell’evoluzione temporale di un sistema elettronico. Gli schemi precedenti su macchine a ioni intrappolati implementavano ogni pezzo di un operatore di eccitazione separatamente, usando molte porte MS per ogni termine. In questo lavoro, gli autori mostrano che forme specifiche della porta MS possono diagonalizzare intere famiglie di questi termini contemporaneamente. Posizionando semplici rotazioni su singoli qubit tra appena due porte MS, riescono ad applicare molti componenti non locali in parallelo. Per le eccitazioni a un singolo elettrone questo dimezza il numero di porte MS richieste, e per le eccitazioni a doppio elettrone le riduce di un fattore quattro, senza bisogno di qubit ausiliari aggiuntivi.
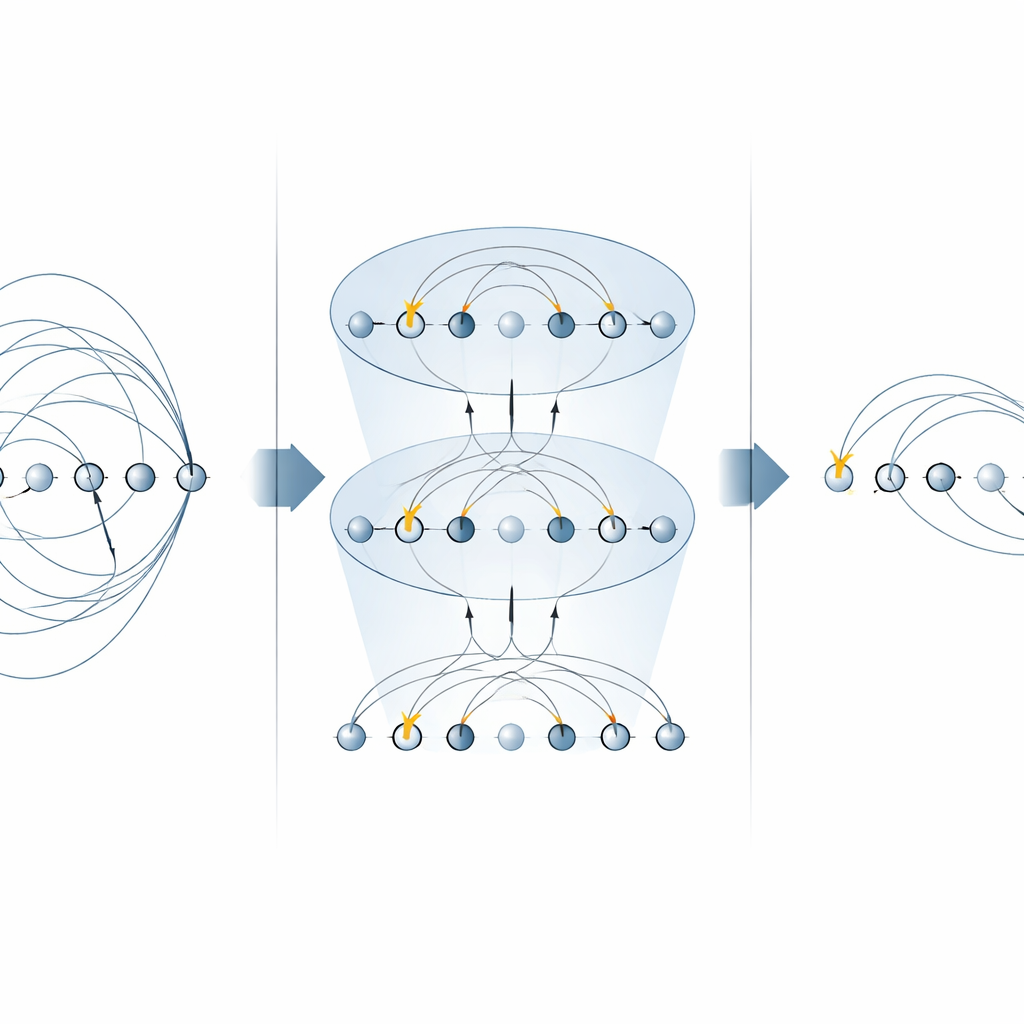
Costruire circuiti di chimica quantistica più veloci
Con questi blocchi ottimizzati, gli autori costruiscono circuiti completi sia per ricerche variazionali dello stato fondamentale sia per la dinamica in tempo reale. Illustrano il metodo su uno ione molecolare piccolo ma non banale, H3+, mostrando come assemblare un intero strato UCCSD (singole e doppie) e un passo di Trotter per l’evoluzione temporale usando molte meno porte globali rispetto agli approcci precedenti. La stessa strategia si generalizza a eccitazioni di ordine superiore, apporta benefici ad ansätze alternativi di «eccitazione su qubit» popolari per dispositivi a breve termine, e può essere riutilizzata per simulare direttamente Hamiltoniane elettroniche. È importante che l’approccio rispetti quantità fisiche chiave come il numero di particelle e lo spin, centrali nelle applicazioni chimiche.
Testare le prestazioni sotto rumore realistico
I circuiti più corti sono utili solo se effettivamente riducono gli errori sull’hardware reale. Per verificarlo, il team ha costruito un modello dettagliato di rumore per una trappola lineare a 12 ioni, includendo fluttuazioni nelle frequenze dei modi vibrazionali e nelle potenze laser—principali sorgenti di errore negli esperimenti odierni. Hanno quindi confrontato i loro nuovi circuiti con quelli standard per una gamma di piccole molecole, monitorando errori di energia, perdita di fedeltà quantistica e violazioni delle quantità conservate. Sia per le eccitazioni singole che doppie, i progetti migliorati hanno ridotto l’infedeltà dei circuiti di circa la metà fino a un ordine di grandezza intero. Per calcoli molecolari completi, hanno costantemente avvicinato le energie simulate e gli osservabili fisici ai risultati ideali, e il loro vantaggio è diventato più pronunciato per eccitazioni più complesse e sistemi di dimensioni maggiori.
Cosa significa per le simulazioni future
Lo studio non sostiene che la chimica quantistica perfetta sia dietro l’angolo; con i livelli di rumore attuali, anche i circuiti migliorati spesso non bastano da soli per superare le migliori approssimazioni classiche. Tuttavia, il lavoro dimostra che adattando gli algoritmi all’hardware—in questo caso allineando la struttura delle eccitazioni fermioniche con le interazioni globali nelle trappole di ioni—si possono ridurre drasticamente gli overhead e migliorare l’accuratezza. Combinando queste strategie con tecniche di mitigazione degli errori e approssimazioni più modeste come le eccitazioni basate su qubit, è possibile che dispositivi a ioni intrappolati a breve termine affrontino problemi chimicamente rilevanti appena oltre la portata dei computer classici.
Citazione: Kaldenbach, T.N., Schultheis, E., Stewen, N. et al. Improved strategies for fermionic quantum simulation with global interactions. npj Quantum Inf 12, 54 (2026). https://doi.org/10.1038/s41534-026-01223-0
Parole chiave: calcolo quantistico con ioni intrappolati, simulazione di fermioni, chimica quantistica molecolare, porte Mølmer–Sørensen, accoppiamento unitario cluster