Clear Sky Science · tr
ALS ve FTD’de somatik mozaikizm, yaygın dejenerasyonla ilişkili odak mutasyonları tanımlıyor
Beyin hücrelerindeki gizli kusurlar
Amiyotrofik lateral skleroz (ALS) ve frontotemporal demans (FTD), insanların hareketini, konuşmasını ve kişiliğini elinden alan yıkıcı hastalıklardır. Hastaların çoğunda aile öyküsü yoktur; bu da sinir hücrelerini çöküş yoluna ilk olarak neyin soktuğu konusunda bir gizem bırakır. Bu çalışma, sadece beyin ve omuriliğin küçük bölgelerinde ortaya çıkan, geç dönemde beliren küçük DNA hatalarının sözde sporadik vakalarda hastalığı tetikleyip tetiklemediğini inceliyor.
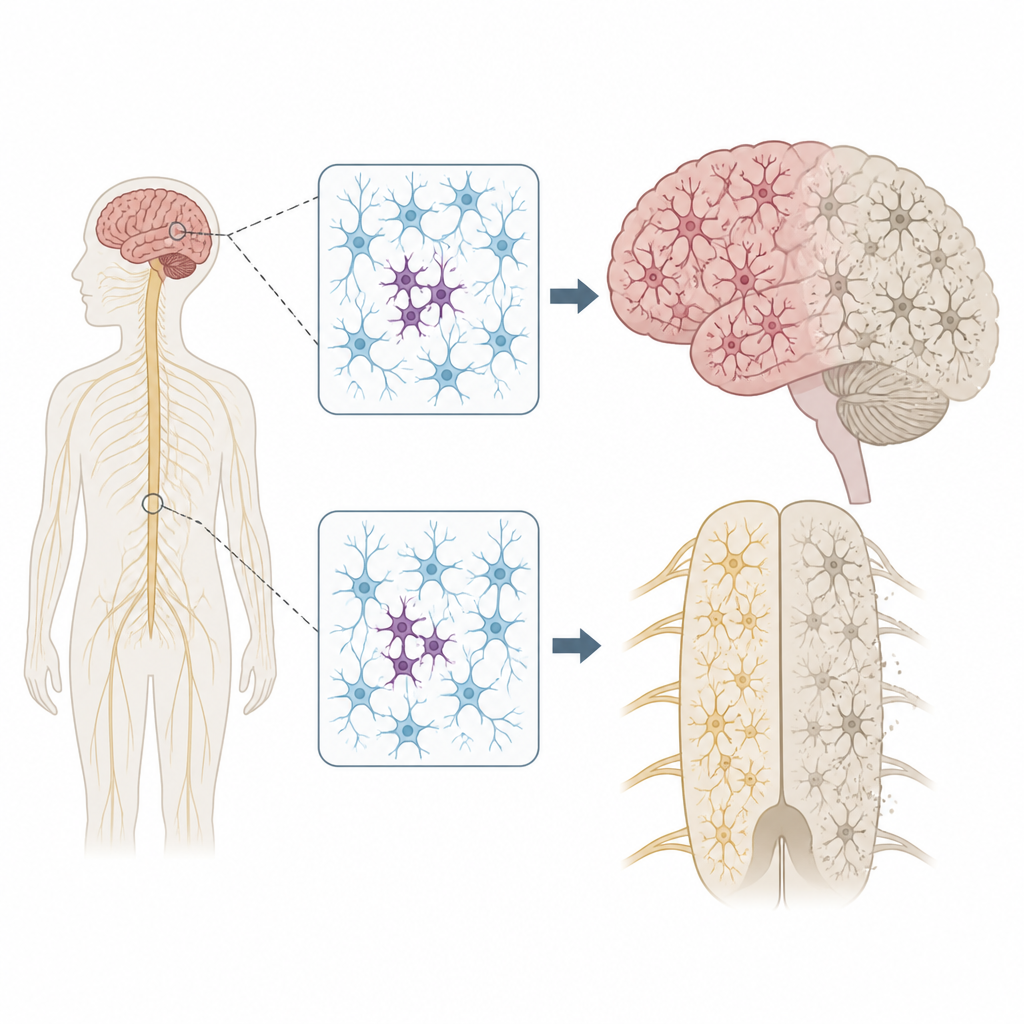
Noktasal DNA değişiklikleri neden önemli
Her insan, hastalık riskini artırabilecek veya azaltabilecek birkaç kalıtsal DNA varyantı taşır. Ancak genomlarımız mükemmel şekilde sabit kalmaz. Hücreler bölünüp yaşlandıkça, yalnızca bazı hücrelerde yeni mutasyonlar ortaya çıkabilir; buna mozaikizm denir. Yazarlar, sinir hücrelerinde böyle yamalı mutasyonların ALS ve FTD’nin genellikle bir vücut bölgesinde —örneğin tek bir el veya dilin bir tarafı— başlayıp sonra sinir sistemi genelinde yayılma eğilimini açıklayıp açıklamayacağını merak ettiler.
Sinir sistemindeki kilit genleri taramak
Bu fikri test etmek için ekip, ölümdən sonra bağışlanmış dokuları inceleyerek 291 ALS, 117 FTD vakası ve nörolojik hastalığı olmayan 144 bireyi değerlendirdi. İstemdışı hareketi kontrol eden primer motor korteks ve motor nöronların bulunduğu omuriliği de içeren birden çok bölgeye odaklandılar. Ultra-derin dizileme yöntemi kullanarak nörodejenerasyonla ilişkili 88 geni binlerce kez okudular; bu, her örnekte yalnızca küçük bir hücre kesrinde bulunan nadir mutasyonları tespit etmelerine olanak sağladı.
Kalıtsal risk ve yeni mutasyonlar
Verilerin ilk geçirilmesi, kalıtsal ve tüm vücutta bulunan gen değişikliklerini aradı. ALS ve FTD vakalarının yaklaşık yüzde 30’u, tanımlı hastalık genlerinde bilinen veya güçlü şekilde tahmin edilen zararlı varyantlar taşıyordu; bu, aile öyküsü olmasa bile genetik testin değerini güçlendiriyor. Araştırmacılar daha sonra bu vakaları analizin geri kalanından çıkartıp germline riski olmayan hastalara odaklandı. Bu kalan grupta, yaklaşık yüzde 2,1’inin yalnızca beyin veya omurilik hücrelerinin bir alt kesrinde ortaya çıkan yeni, zararlı mutasyonlara sahip olduğu bulundu. Bu somatik varyantlar kontrol dokularında neredeyse hiç bulunmuyordu.
Uzak etkileri olan odak sorunlar
Bu yamalı mutasyonların yerleşimi çarpıcıydı. ALS’de, hastalık genlerindeki zararlı varyantlar primer motor korteks ve omurilikte kümeleniyordu; buralar en fazla sinir hücresi kaybının görüldüğü bölgelerdir. Bu mutasyonların birçoğu çok düşük düzeylerde, sıklıkla hücrelerin yüzde 2’sinden daha azında bulunuyordu ve tek bir bölgeyle sınırlıydı; bu da bunların yaşamın geç döneminde ortaya çıktığını düşündürüyor. Yine de çevre dokuda yanlış katlanmış bir protein olan TDP-43’ün yaygın birikimi ve yoğun sinir hücresi kaybı gözleniyordu; bu, kalıtsal mutasyonlu vakalara benzer bir tablo sunuyor. Bu desen, küçük bir mutant hücre grubunun hastalığı lokal olarak başlattığı ve ardından toksik protein değişikliklerinin bağlantılı beyin ile omurilik devreleri boyunca dışa doğru yayıldığı senaryosunu destekliyor.
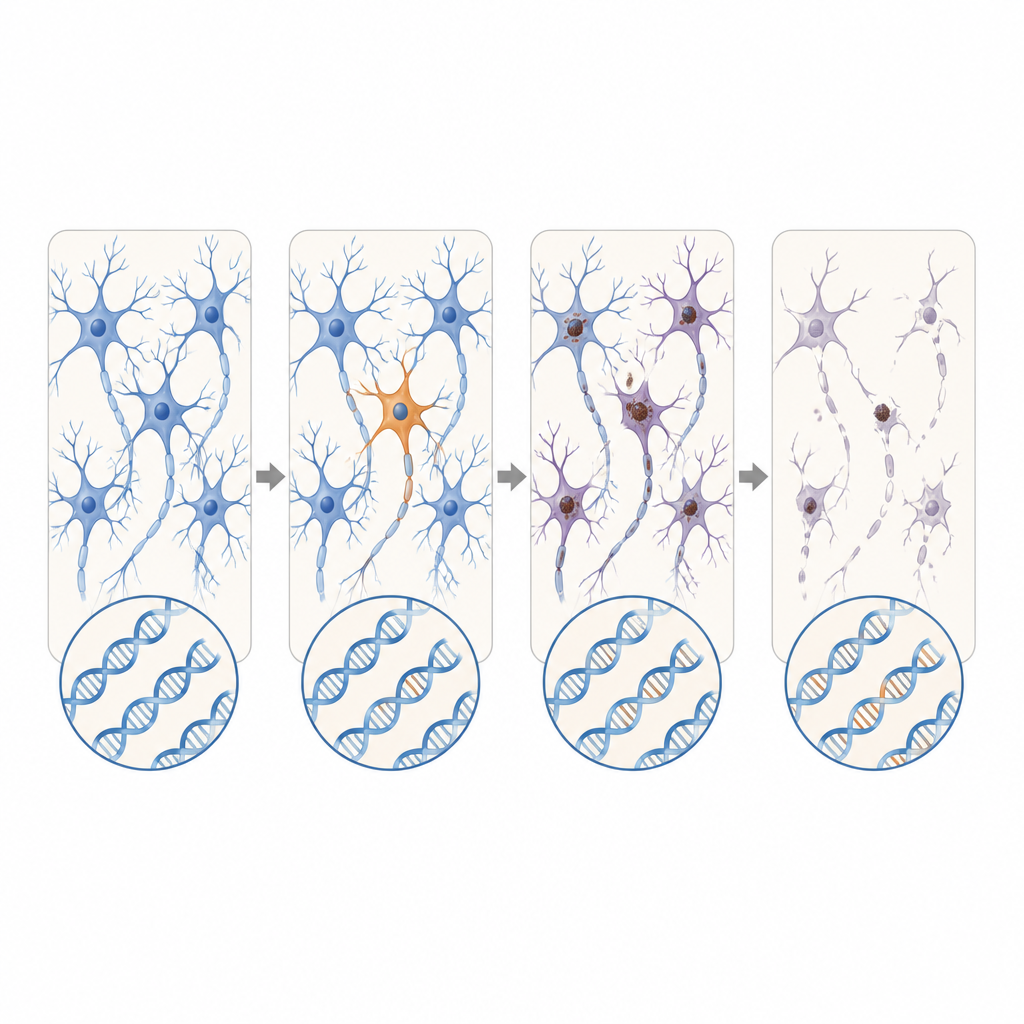
Yeni aday genler ve genişleyen tekrarlar
Bilinen ALS ve FTD genlerinin ötesinde, bilim insanları ek ALS bağışçılarından alınan toplu RNA verilerindeki aktif genler genelinde de arama yaptılar. DYNC1H1 ve LMNA'da zararlı somatik varyantlar ortaya çıkardılar; bu genlerin kalıtsal formları, genellikle ALS’in görüldüğü yaşa ulaşmayı engelleyen ciddi çocukluk motor nöron bozukluklarına yol açar. Bu hastalarda kusurlu versiyonlar yalnızca sinir sistemi hücrelerinin bir bölümünde bulunuyordu; bu durum normal gelişime izin verip daha sonra geç başlangıçlı dejenerasyona yol açmayı mümkün kılabilir. Uzun okuma dizilemesi kullanarak ekip ayrıca, C9orf72 genindeki kısa, görünüşte zararsız bir tekrar dizisinin yalnızca beyin hücrelerinde dramatik şekilde genişlediği ve hastalığa neden olduğu bilinen aralığa geçtiği bir FTD vakasını da tespit etti.
Hastalar ve aileleri için anlamı
Bir arada ele alındığında bulgular, görünüşte sporadik ALS veya FTD olan kişilerin küçük fakat anlamlı bir payı için tetikleyicinin tüm vücutta değil, sinir sisteminin yalnızca ince bir dilimde ortaya çıkan nadir DNA değişiklikleri olabileceğini öne sürüyor. Bu gizli kusurlar standart kan testlerinde çok lokal ve çok ince oldukları için görünmezler, ancak yine de protein yanlış katlanması ve sinir hücresi ölümü zincir reaksiyonunu başlatmaya yeterli olabilir ve sonunda büyük beyin ve omurilik bölgelerini etkileyebilir. Mevcut yöntemler muhtemelen bu tür olayların yalnızca bir kısmını tespit edebildiğinden, bu çalışma mozaik mutasyonları bulmak ve patolojinin bu küçük başlangıç noktalarından nasıl yayıldığını haritalamak için daha hassas araçlara duyulan ihtiyacı işaret ediyor.
Atıf: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Anahtar kelimeler: amiyotrofik lateral skleroz, frontotemporal demans, somatik mozaikizm, nörodejenerasyon, C9orf72 tekrar genişlemesi