Clear Sky Science · fr
Mosaïcisme somatique dans la SLA et la DFT identifie des mutations focales associées à une dégénérescence étendue
Failles cachées dans les cellules cérébrales
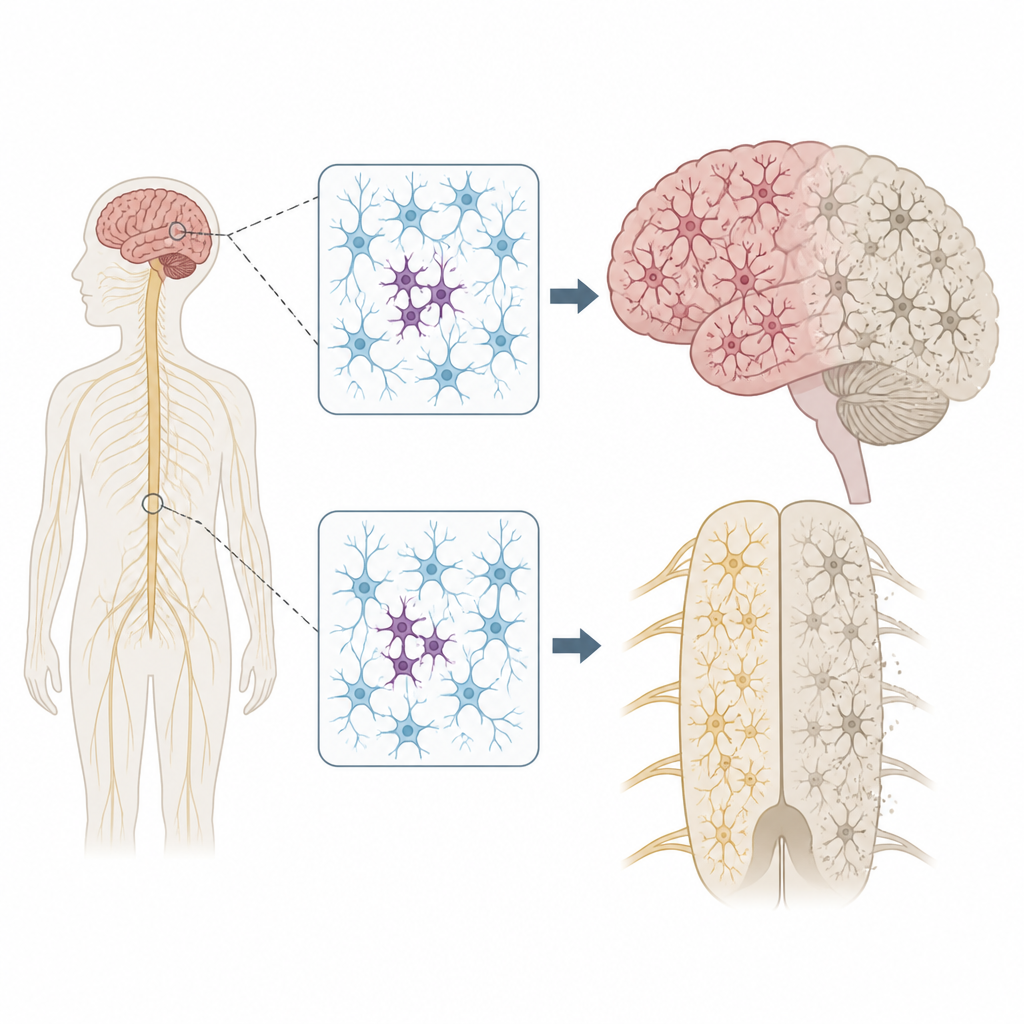
La sclérose latérale amyotrophique (SLA) et la démence frontotemporale (DFT) sont des affections dévastatrices qui privent les personnes de la mobilité, de la parole et de la personnalité. La plupart des patients n’ont pas d’antécédents familiaux, ce qui laisse un mystère sur ce qui met initialement leurs cellules nerveuses sur la voie de l’effondrement. Cette étude s’interroge pour savoir si de minuscules anomalies de l’ADN, survenant tardivement et uniquement dans de petites zones du cerveau et de la moelle épinière, pourraient contribuer à déclencher la maladie dans ces cas dits sporadiques.
Pourquoi des changements d’ADN dispersés comptent
Chaque personne porte quelques variantes d’ADN héritées qui peuvent augmenter ou diminuer le risque de maladie. Mais nos génomes ne restent pas parfaitement fixes. Au fur et à mesure que les cellules se divisent et vieillissent, de nouvelles mutations peuvent apparaître dans seulement certaines cellules, un état appelé mosaïcisme. Les auteurs se sont demandé si de telles mutations parcimonieuses dans les neurones pouvaient aider à expliquer pourquoi la SLA et la DFT commencent souvent dans une région du corps — par exemple une main ou un côté de la langue — avant de se propager plus largement dans le système nerveux.
Analyse des gènes clés du système nerveux
Pour tester cette idée, l’équipe a examiné des tissus donnés après décès provenant de 291 personnes atteintes de SLA, 117 de DFT, et 144 individus sans maladie neurologique. Ils se sont concentrés sur plusieurs régions, dont le cortex moteur primaire, qui contrôle le mouvement volontaire, et la moelle épinière, qui abrite les motoneurones. En utilisant une méthode de séquençage ultra-profond, ils ont lu 88 gènes liés à la neurodégénérescence des milliers de fois, ce qui leur a permis de détecter des mutations rares présentes seulement dans une petite fraction des cellules de chaque échantillon.
Risque hérité et nouvelles mutations
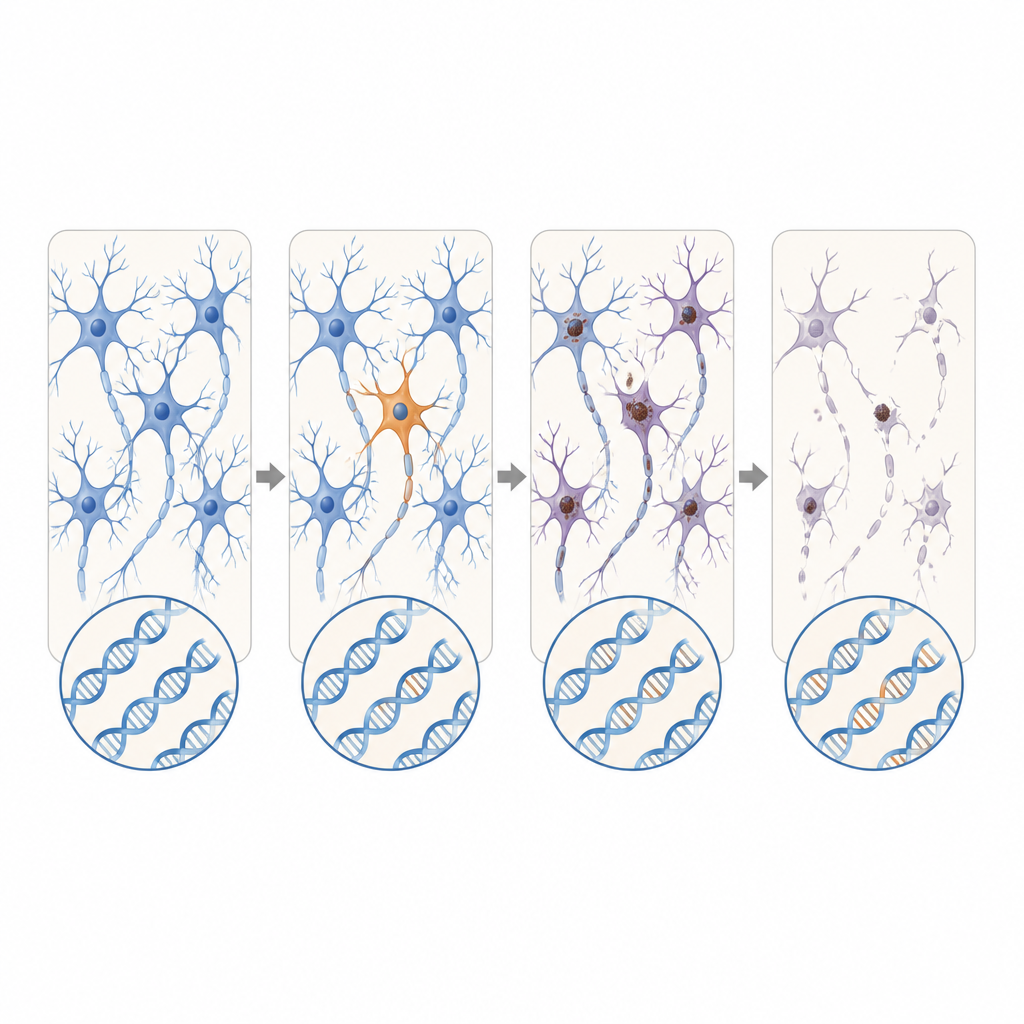
Le premier passage des données a recherché des variations génétiques héritées et présentes dans tout l’organisme. Environ 30 % des cas de SLA et de DFT portaient des variants connus ou fortement prédits comme délétères dans des gènes établis de la maladie, ce qui renforce l’intérêt des tests génétiques même en l’absence d’antécédents familiaux. Les chercheurs ont ensuite écarté ces cas pour se concentrer sur les patients dépourvus de tel risque germinal. Dans ce groupe restant, ils ont trouvé qu’environ 2,1 % portaient de nouvelles mutations délétères n’apparaissant que dans un sous-ensemble de cellules du cerveau ou de la moelle épinière. Ces variants somatiques étaient quasiment absents dans les tissus témoins.
Problèmes focaux aux effets étendus
L’emplacement de ces mutations parcimonieuses était frappant. Dans la SLA, des variants délétères dans des gènes de la maladie s’agglutinaient dans le cortex moteur primaire et la moelle épinière, précisément les régions montrant la plus grande perte neuronale. Nombre de ces mutations étaient présentes à des niveaux extrêmement faibles, souvent chez moins de 2 % des cellules, et confinées à une région, ce qui suggère qu’elles sont apparues tard dans la vie. Pourtant, les tissus environnants montraient une accumulation étendue d’une protéine mal repliée appelée TDP-43 et une forte perte neuronale, à l’image des cas avec mutations héritées. Ce schéma soutient l’hypothèse selon laquelle un petit groupe de cellules mutantes initie localement la maladie, et que des altérations protéiques toxiques se propagent ensuite via des circuits connectés du cerveau et de la moelle épinière.
Nouveaux gènes candidats et répétitions en expansion
Au-delà des gènes connus de la SLA et de la DFT, les chercheurs ont aussi exploré les gènes actifs à partir de données d’ARN en vrac provenant d’autres donneurs atteints de SLA. Ils ont découvert des variants somatiques délétères dans DYNC1H1 et LMNA, des gènes qui, sous forme héritée, provoquent des troubles moteurs infantiles sévères qui empêchent généralement la survie jusqu’à l’âge où la SLA survient. Chez ces patients, les versions défectueuses apparaissaient uniquement dans une portion des cellules du système nerveux, permettant potentiellement un développement normal suivi d’une dégénérescence à apparition tardive. Grâce au séquençage longue lecture, l’équipe a également identifié un cas de DFT dans lequel une courte répétition d’ADN apparemment bénigne dans le gène C9orf72 s’était considérablement élargie uniquement dans les cellules cérébrales, atteignant une fourchette connue pour provoquer la maladie.
Que cela signifie pour les patients et les familles
Pris ensemble, ces résultats suggèrent que, pour une part petite mais significative des personnes atteintes de SLA ou de DFT apparemment sporadiques, le déclencheur peut être des modifications d’ADN rares qui surgissent dans une fine portion du système nerveux plutôt que dans chaque cellule du corps. Ces failles cachées sont trop localisées et trop subtiles pour apparaître dans les analyses sanguines standards, et pourtant elles peuvent suffire à lancer une réaction en chaîne de mauvais repliement protéique et de mort neuronale qui finit par affecter de larges régions du cerveau et de la moelle épinière. Bien que les méthodes actuelles ne puissent probablement détecter qu’une fraction de tels événements, ce travail souligne la nécessité d’outils plus sensibles pour repérer les mutations mosaïques et cartographier comment la pathologie se propage depuis ces tout petits points de départ.
Citation: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Mots-clés: Sclérose latérale amyotrophique, Démence frontotemporale, mosaïcisme somatique, neurodégénérescence, expansion de répétition C9orf72