Clear Sky Science · sv
Somatisk mosaik i ALS och FTD identifierar fokala mutationer kopplade till omfattande degeneration
Dolda fel i hjärnceller
Amyotrofisk lateralskleros (ALS) och frontotemporal demens (FTD) är förödande sjukdomar som berövar människor rörelse, tal och personlighet. De flesta patienter har ingen familjehistoria, vilket lämnar en gåta kring vad som först får deras nervceller att gå mot sammanbrott. Denna studie undersöker om små, sena DNA-fel som uppstår endast i små områden av hjärnan och ryggmärgen kan hjälpa till att utlösa sjukdomen i dessa så kallade sporadiska fall.
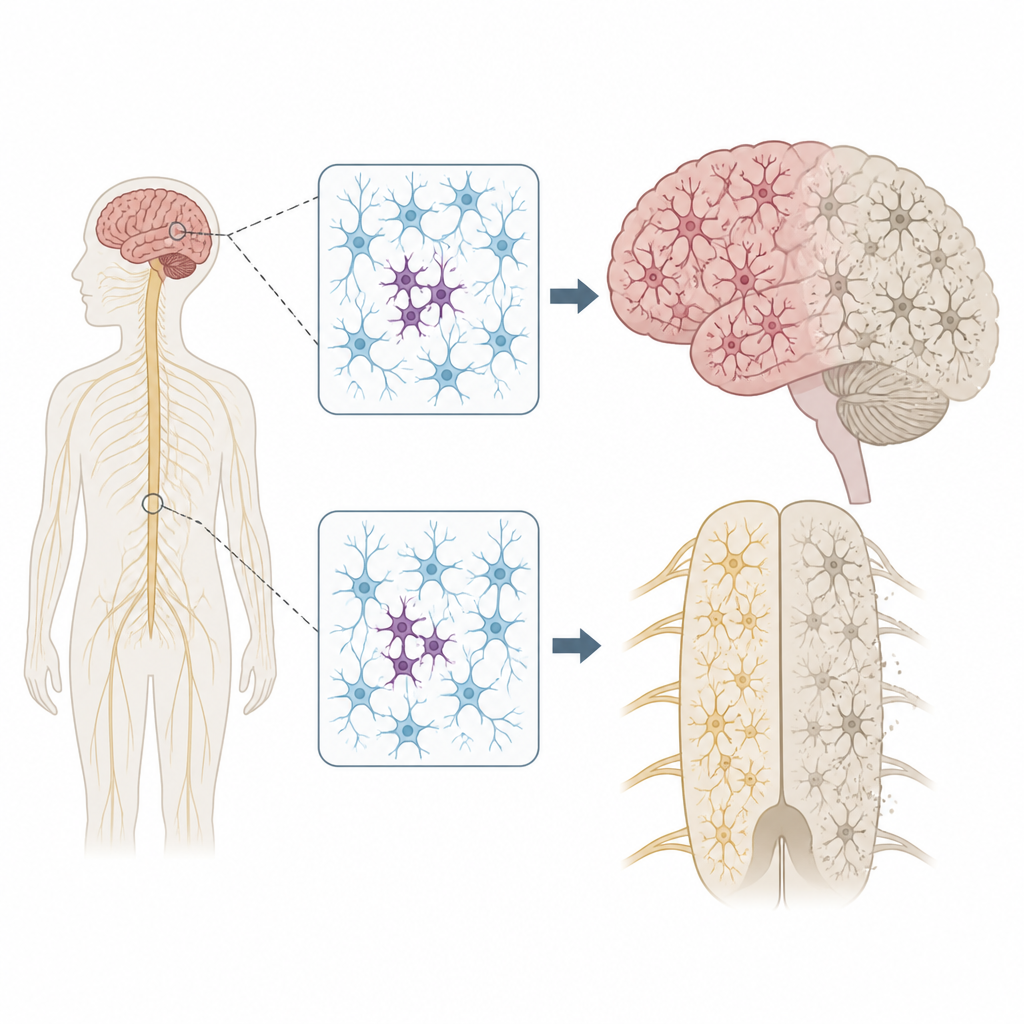
Varför spridda DNA-förändringar spelar roll
Varje person bär på några ärftliga DNA-varianter som kan höja eller sänka risken för sjukdom. Men våra genom förblir inte helt oförändrade. När celler delar sig och åldras kan nya mutationer uppstå endast i vissa celler, ett tillstånd som kallas mosaik. Författarna undrade om sådana fläckvisa mutationer i nervceller kan hjälpa till att förklara varför ALS och FTD ofta börjar i en kroppsregion, till exempel en hand eller ena sidan av tungan, innan de sprider sig mer allmänt genom nervsystemet.
Skanning av nyckelgener i nervsystemet
För att testa idén undersökte teamet vävnad skänkt efter döden från 291 personer med ALS, 117 med FTD och 144 individer utan neurologisk sjukdom. De fokuserade på flera regioner, inklusive primära motorcortex, som styr viljemässig rörelse, och ryggmärgen, som rymmer motorneuron. Med en ultradjup sekvenseringsmetod läste de 88 gener kopplade till neurodegeneration tusentals gånger, vilket gjorde det möjligt att upptäcka sällsynta mutationer som fanns endast i en liten andel celler i varje prov.
Ärftlig risk och nya mutationer
Första genomgången av data sökte efter ärftliga, kroppsomfattande genförändringar. Ungefär 30 procent av ALS- och FTD-fallen bar på kända eller starkt förmodade skadliga varianter i etablerade sjukdomsgener, vilket förstärker värdet av genetisk testning även när det saknas familjehistoria. Forskarna uteslöt sedan dessa fall från vidare analys för att fokusera på patienter utan sådan germline-risk. I denna återstående grupp fann de att cirka 2,1 procent bar nya, skadliga mutationer som endast verkade finnas i en delmängd av hjärn- eller ryggmärgsceller. Dessa somatiska varianter var nästan helt frånvarande i kontrollvävnader.
Fokala problem med långtgående effekter
Platsen för dessa fläckvisa mutationer var slående. Vid ALS klustrade skadliga varianter i sjukdomsgener i primära motorcortex och ryggmärgen, just de regioner som uppvisar mest nervcellsförlust. Många av dessa mutationer fanns på extremt låga nivåer, ofta i färre än 2 procent av cellerna, och var begränsade till en region, vilket tyder på att de uppstod sent i livet. Trots detta visade omkringliggande vävnad omfattande ansamling av ett missvecklat protein kallat TDP-43 och kraftig nervcellsförlust, likt fallen med ärftliga mutationer. Detta mönster stödjer en scenariouppfattning där en liten grupp mutanta celler startar sjukdomen lokalt, och därefter sprider sig toxiska proteinförändringar utåt genom sammankopplade kretsar i hjärna och ryggmärg.
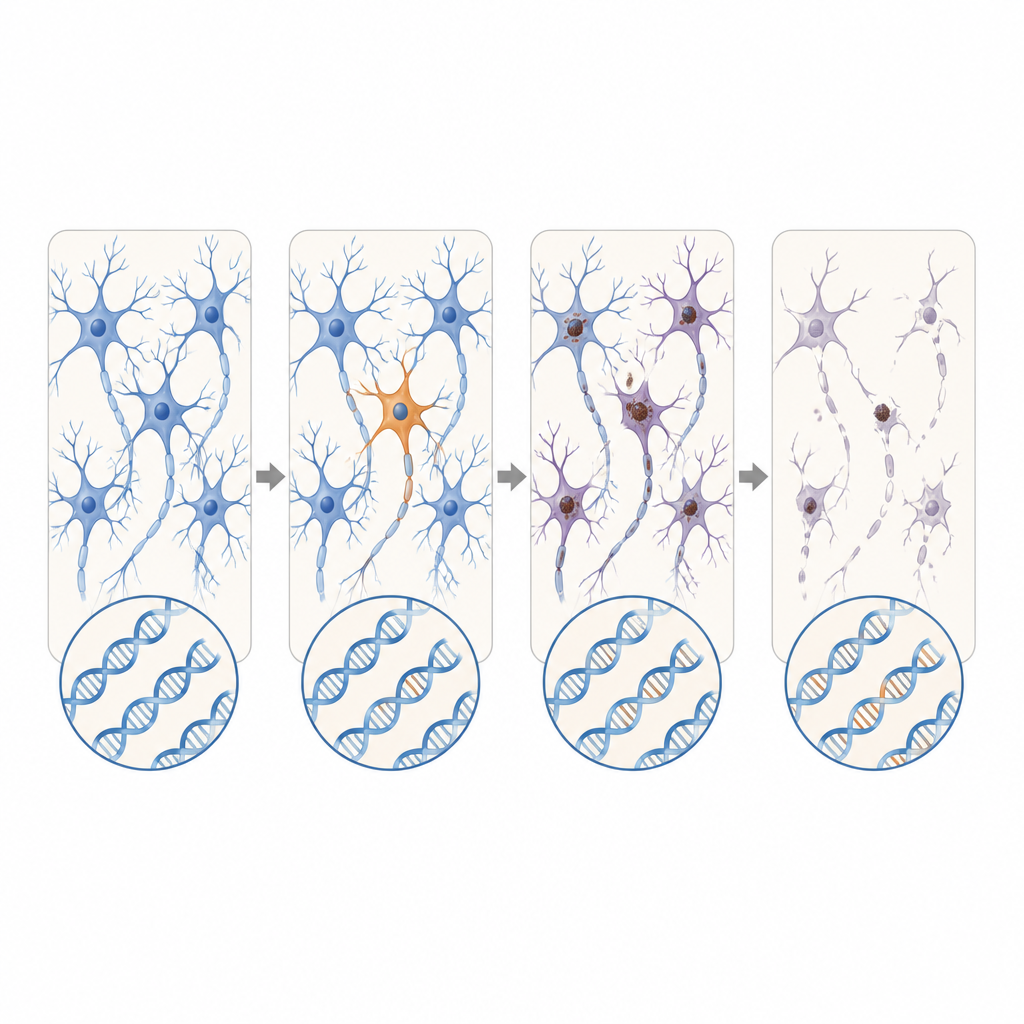
Nya kandidatgener och expanderande upprepningar
Utöver kända ALS- och FTD-gener sökte forskarna också över de aktiva generna i bulk-RNA-data från ytterligare ALS-donatorer. De upptäckte skadliga somatiska varianter i DYNC1H1 och LMNA, gener som i deras ärftliga form orsakar svåra barndomssjukdomar i motorneuron som vanligtvis hindrar överlevnad till den ålder då ALS uppträder. Hos dessa patienter förekom de felaktiga varianterna endast i en del av nervsystemets celler, vilket potentiellt tillät normal utveckling följd av sen debuterande degeneration. Med långläsningssekvensering identifierade teamet också ett FTD-fall där en kort, till synes ofarlig sträcka av upprepad DNA i C9orf72-genen hade expanderat dramatiskt endast i hjärnceller och överskridit ett intervall känt för att orsaka sjukdom.
Vad detta betyder för patienter och familjer
Sammantaget tyder fynden på att för en liten men betydelsefull andel av personer med till synes sporadisk ALS eller FTD kan utlösaren vara sällsynta DNA-förändringar som uppstår i bara en smal del av nervsystemet snarare än i varje cell i kroppen. Dessa dolda fel är för lokala och för subtila för att upptäckas i standardprov från blod, men kan ändå räcka för att starta en kedjereaktion av proteinfelveckning och nervcellsdöd som så småningom påverkar stora delar av hjärna och ryggmärg. Även om nuvarande metoder sannolikt bara kan upptäcka en del av sådana händelser, pekar detta arbete på behovet av känsligare verktyg för att hitta mosaikmutationer och kartlägga hur patologin sprider sig från dessa små startpunkter.
Citering: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Nyckelord: amyotrofisk lateralskleros, frontotemporal demens, somatisk mosaik, neurodegeneration, C9orf72-upprepningsexpansion