Clear Sky Science · en
Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration
Hidden faults in brain cells
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating conditions that rob people of movement, speech, and personality. Most patients have no family history, leaving a mystery about what first sets their nerve cells on a path to failure. This study asks whether tiny, late-appearing DNA glitches that arise only in small patches of the brain and spinal cord might help spark disease in these so-called sporadic cases.
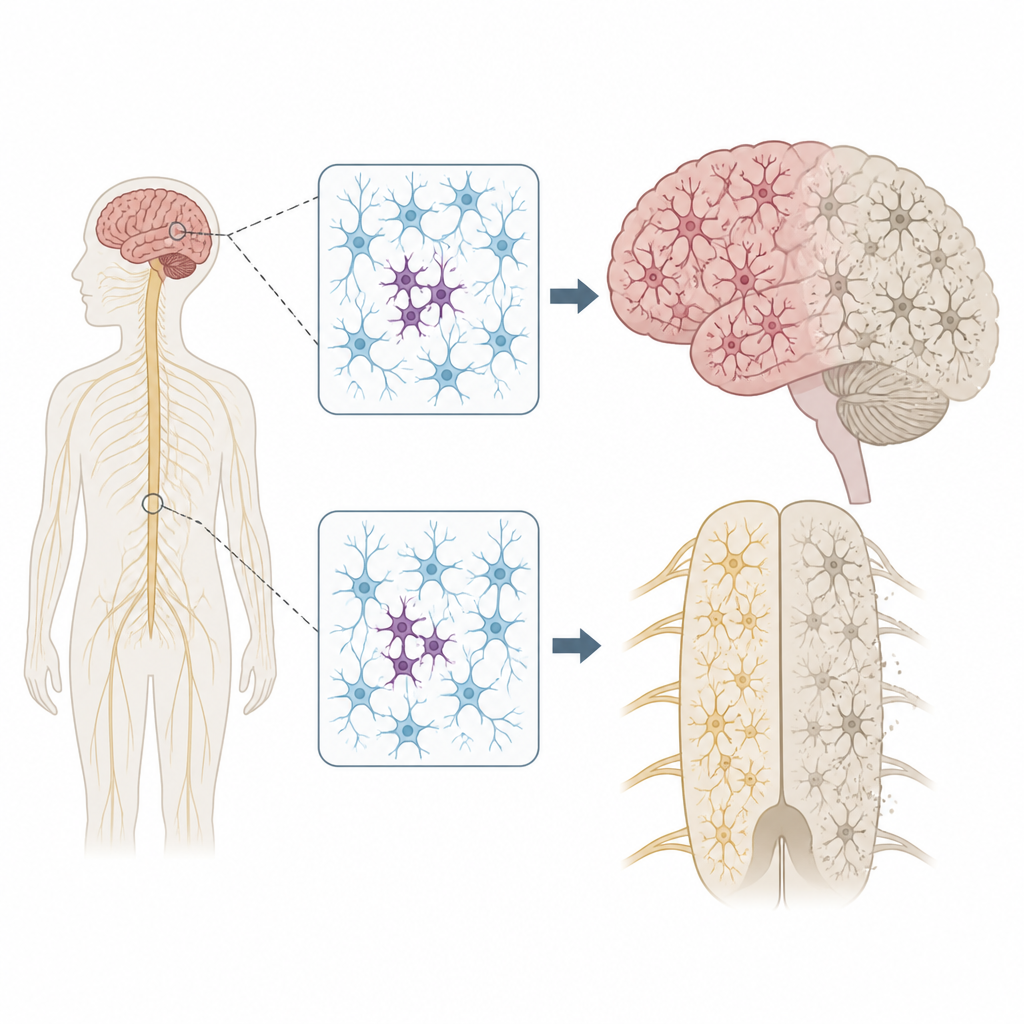
Why scattered DNA changes matter
Every person carries a few inherited DNA variants that can raise or lower disease risk. But our genomes do not stay perfectly fixed. As cells divide and age, new mutations can appear in only some cells, a state called mosaicism. The authors wondered if such patchy mutations in nerve cells could help explain why ALS and FTD often begin in one body region, such as a single hand or one side of the tongue, before spreading more widely through the nervous system.
Scanning key genes in the nervous system
To test this idea, the team examined tissue donated after death from 291 people with ALS, 117 with FTD, and 144 individuals without neurological disease. They focused on multiple regions, including the primary motor cortex, which controls voluntary movement, and the spinal cord, which houses motor neurons. Using an ultra-deep sequencing method, they read 88 genes linked to neurodegeneration thousands of times over, which allowed them to detect rare mutations present in only a small fraction of cells in each sample.
Inherited risk and new mutations
The first pass through the data looked for inherited, body-wide gene changes. Roughly 30 percent of ALS and FTD cases carried known or strongly predicted harmful variants in established disease genes, reinforcing the value of genetic testing even when there is no family history. The researchers then removed those cases from further analysis to focus on patients who lacked such germline risk. In this remaining group, they found that about 2.1 percent harbored new, damaging mutations that appeared only in a subset of brain or spinal cord cells. These somatic variants were almost entirely absent in control tissues.
Focal problems with far-reaching effects
The location of these patchy mutations was striking. In ALS, damaging variants in disease genes clustered in the primary motor cortex and spinal cord, the very regions that show the most nerve cell loss. Many of these mutations were present at extremely low levels, often in fewer than 2 percent of cells, and were confined to one region, suggesting they arose late in life. Yet the surrounding tissue still showed widespread buildup of a misfolded protein called TDP-43 and heavy nerve cell loss, much like cases with inherited mutations. This pattern supports a scenario in which a small group of mutant cells starts the disease locally, and toxic protein changes then spread outward through connected brain and spinal cord circuits.
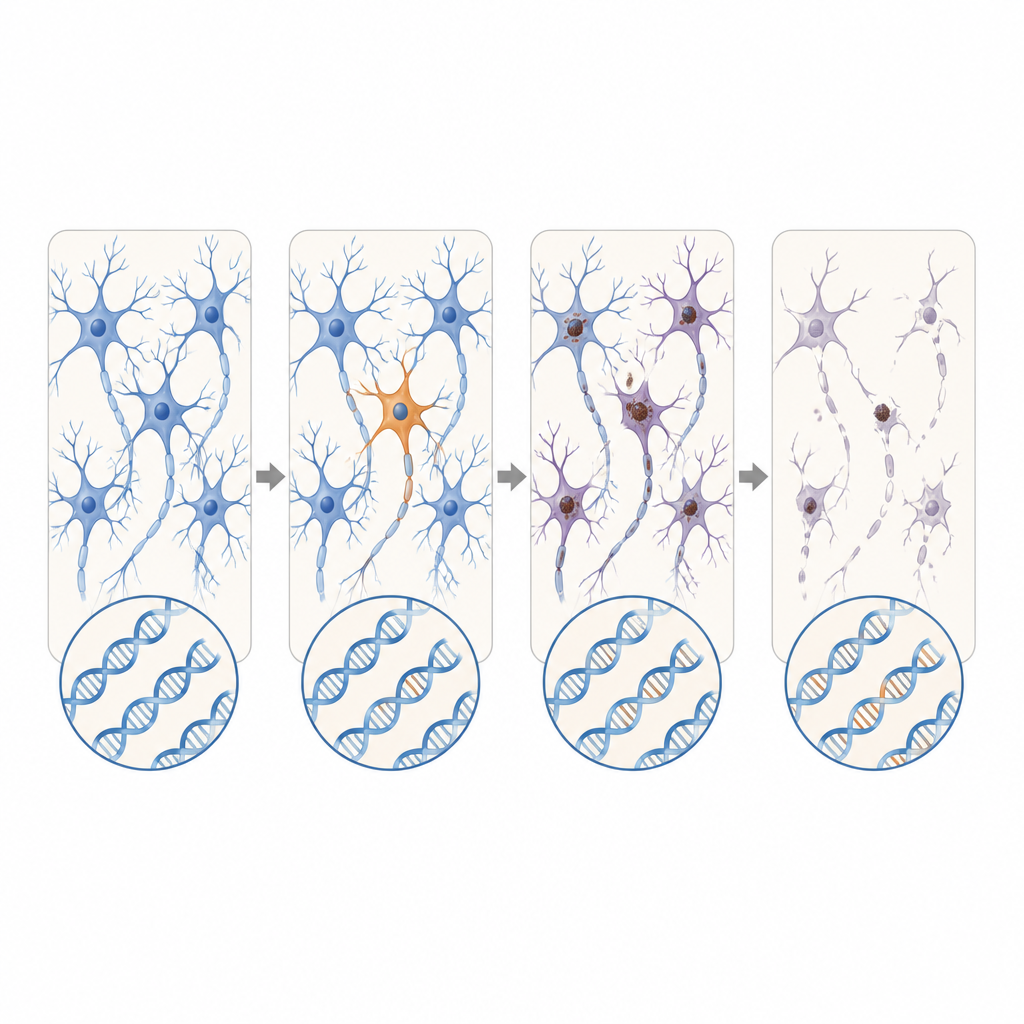
New candidate genes and expanding repeats
Beyond known ALS and FTD genes, the scientists also searched across the active genes in bulk RNA data from additional ALS donors. They uncovered damaging somatic variants in DYNC1H1 and LMNA, genes that in their inherited form cause severe childhood motor neuron disorders that usually prevent survival into the age when ALS strikes. In these patients, the faulty versions appeared in only a portion of nervous system cells, potentially allowing normal development followed by late-onset degeneration. Using long-read sequencing, the team also identified an FTD case in which a short, apparently harmless stretch of repeated DNA in the C9orf72 gene had expanded dramatically only in brain cells, crossing into a range known to cause disease.
What this means for patients and families
Taken together, the findings suggest that, for a small but meaningful share of people with apparently sporadic ALS or FTD, the trigger may be rare DNA changes that arise in just a sliver of the nervous system rather than in every cell of the body. These hidden faults are too local and too subtle to show up in standard blood tests, yet they may be enough to start a chain reaction of protein misfolding and nerve cell death that eventually affects large brain and spinal cord regions. Although current methods can probably detect only a fraction of such events, this work points to the need for more sensitive tools to find mosaic mutations and to map how pathology spreads from these tiny starting points.
Citation: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, somatic mosaicism, neurodegeneration, C9orf72 repeat expansion