Clear Sky Science · it
Mosaico somatico nella SLA e FTD identifica mutazioni focali associate a degenerazione diffusa
Difetti nascosti nelle cellule cerebrali
La sclerosi laterale amiotrofica (SLA) e la demenza frontotemporale (FTD) sono condizioni devastanti che privano le persone del movimento, del linguaggio e della personalità. La maggior parte dei pazienti non presenta una storia familiare, lasciando irrisolta la domanda su cosa inneschi inizialmente il cedimento delle loro cellule nervose. Questo studio si chiede se piccole, tardive alterazioni del DNA che insorgono solo in porzioni limitate del cervello e del midollo spinale possano contribuire ad avviare la malattia in questi casi cosiddetti sporadici.
Perché le variazioni disperse del DNA contano
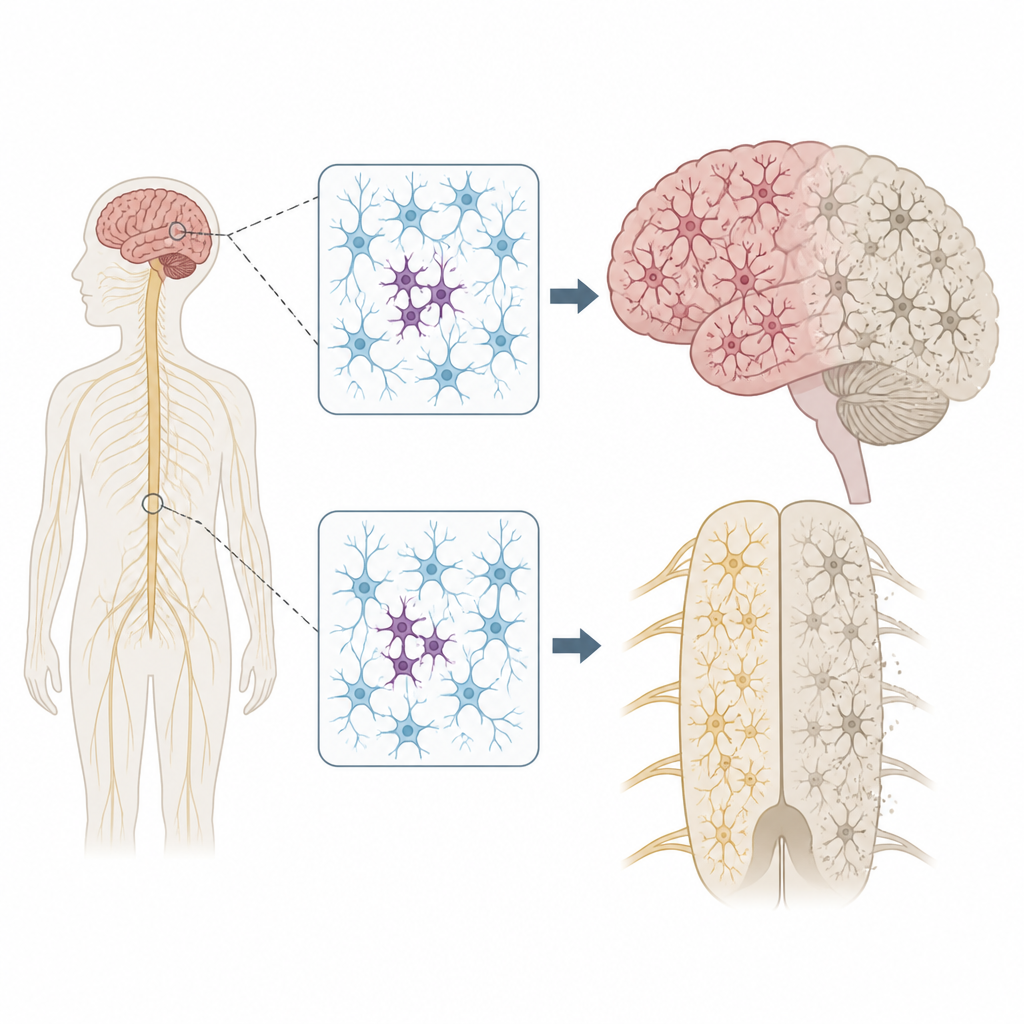
Ogni persona porta con sé alcune varianti del DNA ereditate che possono aumentare o ridurre il rischio di malattia. Ma i nostri genomi non restano perfettamente immutati. Man mano che le cellule si dividono e invecchiano, possono apparire nuove mutazioni solo in alcune cellule, uno stato definito mosaicismo. Gli autori si sono chiesti se tali mutazioni a macchia di leopardo nelle cellule nervose possano aiutare a spiegare perché la SLA e la FTD spesso iniziano in una singola area del corpo, come una mano o un lato della lingua, prima di diffondersi più ampiamente nel sistema nervoso.
Scansione dei geni chiave nel sistema nervoso
Per mettere alla prova questa idea, il gruppo ha analizzato tessuti donati dopo la morte da 291 persone con SLA, 117 con FTD e 144 individui senza malattie neurologiche. Si sono concentrati su più regioni, incluso la corteccia motoria primaria, che controlla il movimento volontario, e il midollo spinale, che ospita i motoneuroni. Utilizzando un metodo di sequenziamento ultra-profondità, hanno letto 88 geni collegati alla neurodegenerazione migliaia di volte, il che ha permesso di rilevare rare mutazioni presenti solo in una piccola frazione di cellule in ciascun campione.
Rischio ereditario e nuove mutazioni
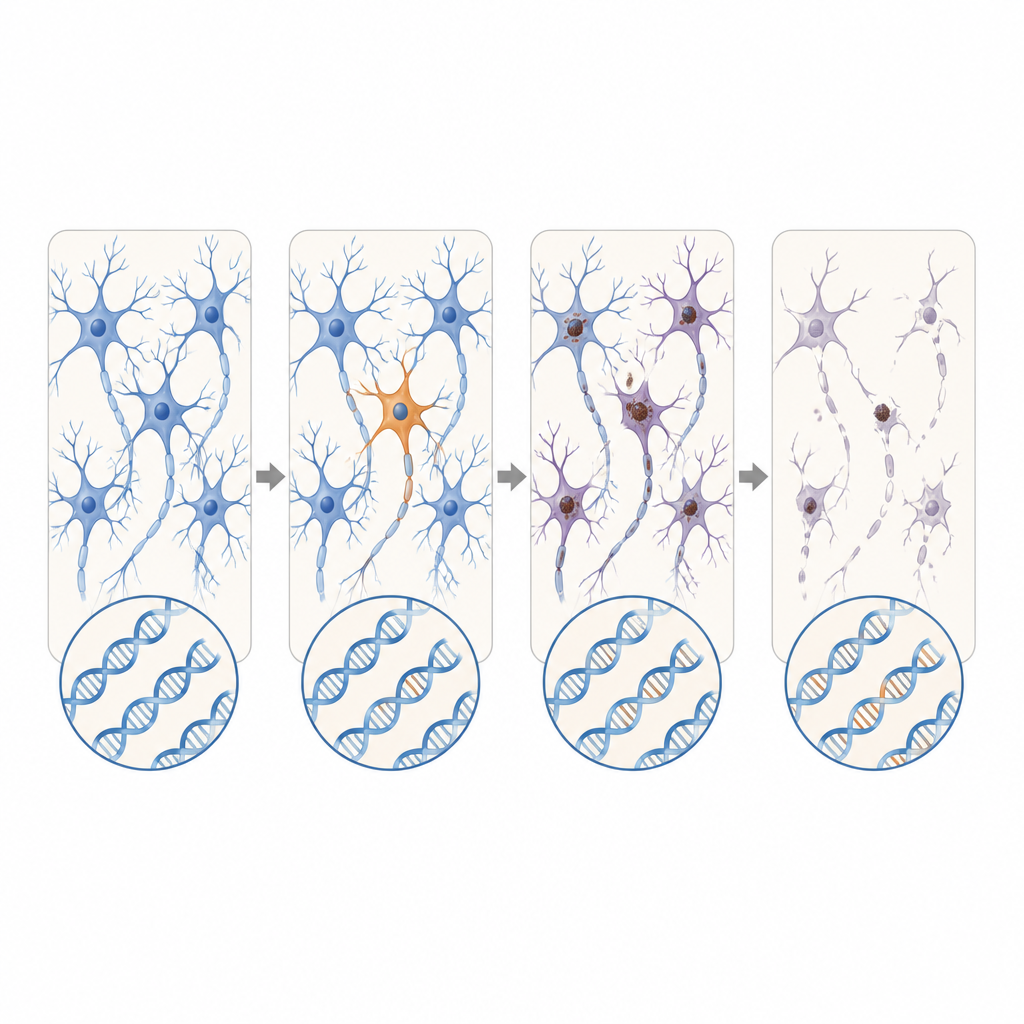
Il primo esame dei dati ha cercato variazioni genetiche ereditate, presenti in tutto il corpo. Circa il 30 percento dei casi di SLA e FTD portava varianti note o fortemente predette come dannose in geni associati alla malattia, rafforzando il valore dei test genetici anche quando non c’è una storia familiare. I ricercatori hanno poi escluso questi casi per concentrarsi su pazienti privi di tale rischio germinale. In questo gruppo rimanente hanno trovato che circa il 2,1 percento presentava nuove mutazioni dannose apparse solo in una sotto-popolazione di cellule del cervello o del midollo spinale. Queste varianti somatiche erano quasi del tutto assenti nei controlli.
Problemi focali con effetti di ampia portata
La posizione di queste mutazioni a macchia di leopardo era notevole. Nella SLA, varianti dannose in geni della malattia si raggruppavano nella corteccia motoria primaria e nel midollo spinale, le stesse regioni che mostrano la maggiore perdita di cellule nervose. Molte di queste mutazioni erano presenti a livelli estremamente bassi, spesso in meno del 2 percento delle cellule, e confinate a una sola regione, suggerendo che siano comparsi tardivamente nella vita. Eppure il tessuto circostante mostrava comunque un ampio accumulo di una proteina malripiegata chiamata TDP-43 e una massiccia perdita di neuroni, molto simile ai casi con mutazioni ereditarie. Questo schema supporta lo scenario secondo cui un piccolo gruppo di cellule mutate innesca la malattia localmente e i cambiamenti proteici tossici si propagano poi attraverso circuiti connessi del cervello e del midollo spinale.
Nuovi geni candidati ed espansioni di ripetizioni
Oltre ai geni noti di SLA e FTD, gli scienziati hanno anche cercato tra i geni espressi nei dati di RNA bulk di ulteriori donatori con SLA. Hanno individuato varianti somatiche dannose in DYNC1H1 e LMNA, geni che nella loro forma ereditaria causano gravi disturbi dei motoneuroni dell’infanzia che solitamente non consentono la sopravvivenza fino all’età in cui insorge la SLA. In questi pazienti, le versioni difettose comparivano solo in una porzione delle cellule del sistema nervoso, consentendo potenzialmente uno sviluppo normale seguito da degenerazione a esordio tardivo. Utilizzando il sequenziamento a lettura lunga, il team ha anche identificato un caso di FTD in cui un breve tratto apparentemente innocuo di ripetizioni nel gene C9orf72 si era espanso in modo drammatico solo nelle cellule cerebrali, raggiungendo una soglia nota per causare malattia.
Cosa significa per pazienti e famiglie
Nel complesso, i risultati suggeriscono che, per una quota piccola ma significativa di persone con SLA o FTD apparentemente sporadiche, il fattore scatenante potrebbe essere costituito da rare variazioni del DNA che insorgono in una sola porzione del sistema nervoso anziché in ogni cellula del corpo. Questi difetti nascosti sono troppo localizzati e troppo sottili per comparire nei test standard sul sangue, eppure possono essere sufficienti ad avviare una reazione a catena di malripiegamento proteico e morte neuronale che alla fine interessa ampie aree del cervello e del midollo spinale. Sebbene i metodi attuali possano rilevare probabilmente solo una frazione di tali eventi, questo lavoro sottolinea la necessità di strumenti più sensibili per individuare le mutazioni mosaicche e per mappare come la patologia si diffonde da questi minuscoli punti di partenza.
Citazione: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Parole chiave: sclerosi laterale amiotrofica, demenza frontotemporale, mosaico somatico, neurodegenerazione, espansione di ripetizione C9orf72