Clear Sky Science · es
Mosaicismo somático en ELA y DFT identifica mutaciones focales asociadas con degeneración generalizada
Fallos ocultos en las células cerebrales
La esclerosis lateral amiotrófica (ELA) y la demencia frontotemporal (DFT) son afecciones devastadoras que privan a las personas de la movilidad, el habla y la personalidad. La mayoría de los pacientes no tiene antecedentes familiares, lo que deja en el aire el misterio de qué inicia el deterioro de sus neuronas. Este estudio plantea si pequeñas alteraciones del ADN que aparecen tarde y solo en parches limitados del cerebro y la médula espinal podrían contribuir a desencadenar la enfermedad en estos casos llamados esporádicos.
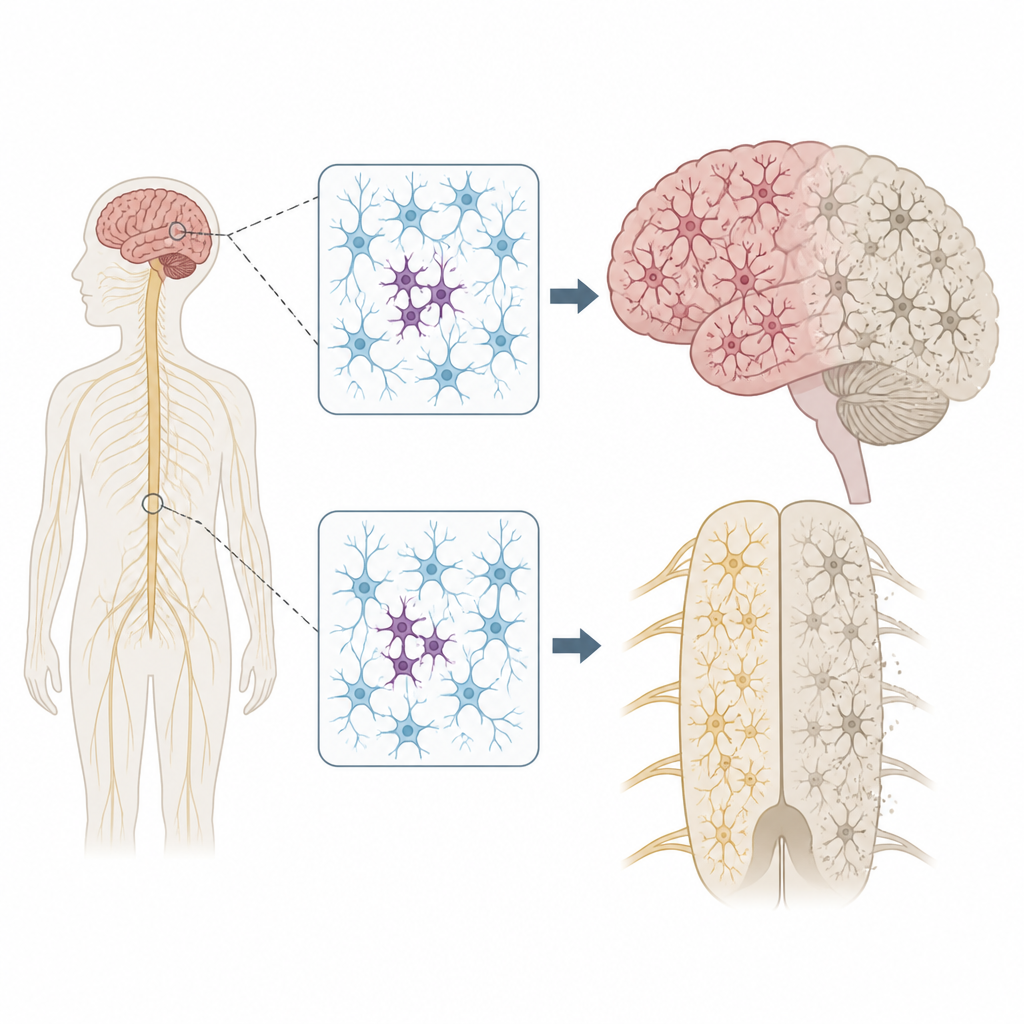
Por qué importan los cambios de ADN dispersos
Cada persona porta algunas variantes hereditarias que pueden aumentar o disminuir el riesgo de enfermedad. Pero nuestros genomas no permanecen inmutables. A medida que las células se dividen y envejecen, pueden surgir nuevas mutaciones en solo algunas células, un estado denominado mosaicismo. Los autores se preguntaron si tales mutaciones parcheadas en las células nerviosas podrían ayudar a explicar por qué la ELA y la DFT a menudo comienzan en una región corporal, como una sola mano o un lado de la lengua, antes de propagarse más ampliamente por el sistema nervioso.
Explorando genes clave en el sistema nervioso
Para poner a prueba esta idea, el equipo examinó tejido donado post mortem de 291 personas con ELA, 117 con DFT y 144 individuos sin enfermedad neurológica. Se centraron en varias regiones, incluida la corteza motora primaria, que controla el movimiento voluntario, y la médula espinal, que alberga las neuronas motoras. Usando un método de secuenciación ultra profunda, leyeron 88 genes vinculados a la neurodegeneración miles de veces, lo que les permitió detectar mutaciones raras presentes en solo una pequeña fracción de células en cada muestra.
Riesgo hereditario y nuevas mutaciones
La primera revisión de los datos buscó cambios genéticos hereditarios presentes en todo el organismo. Aproximadamente el 30 por ciento de los casos de ELA y DFT portaban variantes conocidas o fuertemente predictivas de daño en genes asociados a la enfermedad, lo que refuerza el valor de las pruebas genéticas incluso cuando no hay antecedentes familiares. Los investigadores eliminaron entonces esos casos del análisis para centrarse en pacientes que carecían de tal riesgo germinal. En este grupo restante, encontraron que alrededor del 2,1 por ciento albergaba nuevas mutaciones dañinas que aparecían solo en una subpoblación de células del cerebro o la médula espinal. Estas variantes somáticas estaban prácticamente ausentes en los tejidos control.
Problemas focales con efectos de gran alcance
La localización de estas mutaciones parcheadas fue llamativa. En la ELA, las variantes dañinas en genes de la enfermedad se agruparon en la corteza motora primaria y la médula espinal, las mismas regiones que muestran la mayor pérdida neuronal. Muchas de estas mutaciones estaban presentes en niveles extremadamente bajos, con frecuencia en menos del 2 por ciento de las células, y se limitaban a una región, lo que sugiere que surgieron en etapas tardías de la vida. Sin embargo, el tejido circundante mostraba aun así una acumulación generalizada de una proteína mal plegada llamada TDP-43 y una marcada pérdida neuronal, de forma similar a los casos con mutaciones hereditarias. Este patrón apoya un escenario en el que un pequeño grupo de células mutantes inicia la enfermedad localmente y los cambios proteicos tóxicos se propagan después a través de circuitos conectados del cerebro y la médula espinal.
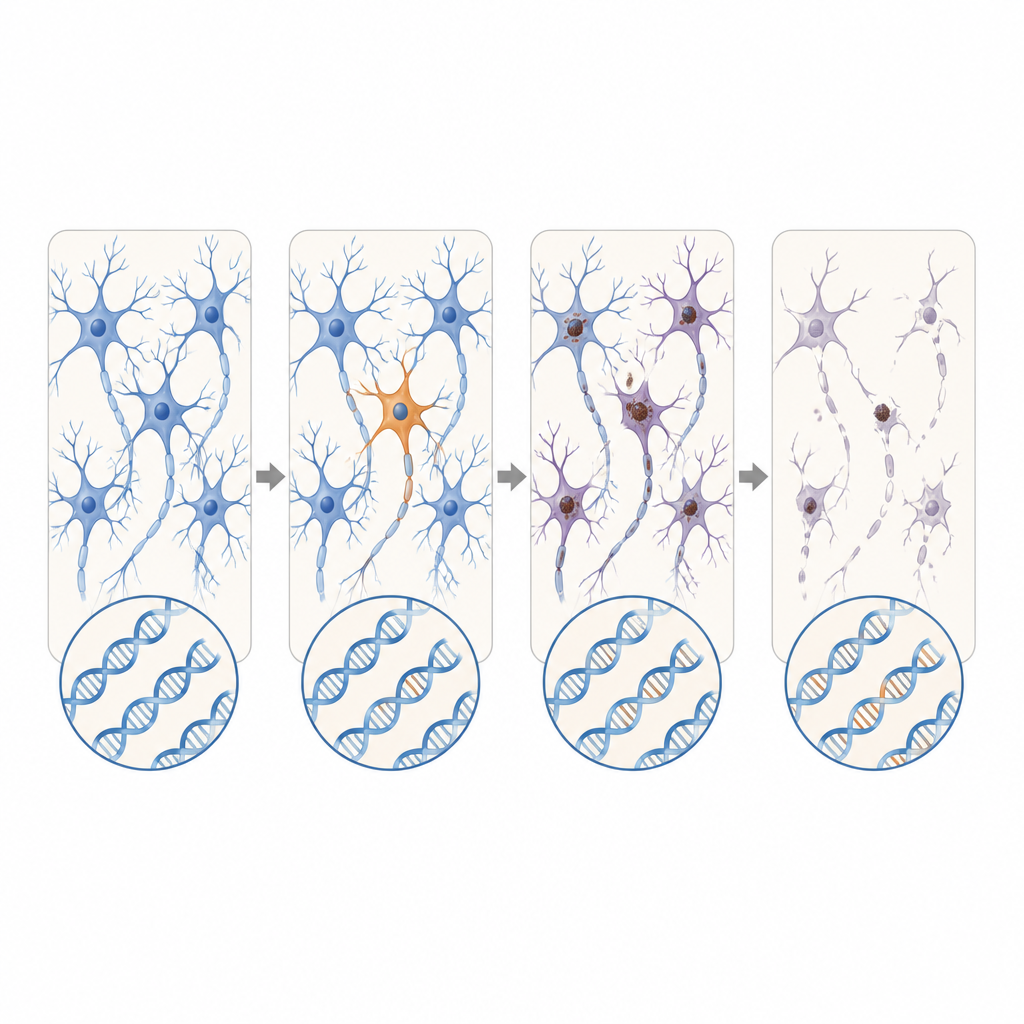
Nuevos genes candidatos y repeticiones en expansión
Más allá de los genes conocidos de ELA y DFT, los científicos también buscaron a lo largo de los genes activos en datos de ARN a granel de donantes adicionales con ELA. Descubrieron variantes somáticas dañinas en DYNC1H1 y LMNA, genes que en su forma heredada causan trastornos motores infantiles severos que suelen impedir la supervivencia hasta la edad en que aparece la ELA. En estos pacientes, las versiones defectuosas aparecían solo en una fracción de las células del sistema nervioso, lo que podría permitir un desarrollo normal seguido de una degeneración de inicio tardío. Mediante secuenciación de lecturas largas, el equipo también identificó un caso de DFT en el que un tramo corto, aparentemente inocuo, de repeticiones en el gen C9orf72 se había expandido de forma dramática solo en células cerebrales, alcanzando un rango conocido por causar enfermedad.
Qué significa esto para pacientes y familias
En conjunto, los hallazgos sugieren que, para una porción pequeña pero relevante de personas con ELA o DFT aparentemente esporádicas, el desencadenante puede ser cambios raros en el ADN que surgen en solo una franja del sistema nervioso en lugar de en todas las células del cuerpo. Estos fallos ocultos son demasiado locales y sutiles para aparecer en pruebas sanguíneas estándar, pero pueden ser suficientes para iniciar una reacción en cadena de mal plegamiento proteico y muerte neuronal que eventualmente afecta amplias regiones del cerebro y la médula espinal. Aunque los métodos actuales probablemente solo detectan una fracción de tales eventos, este trabajo subraya la necesidad de herramientas más sensibles para encontrar mutaciones en mosaico y para mapear cómo se propaga la patología desde estos diminutos puntos de inicio.
Cita: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Palabras clave: esclerosis lateral amiotrófica, demencia frontotemporal, mosaicismo somático, neurodegeneración, expansión de repeticiones en C9orf72