Clear Sky Science · nl
Somatische mosaïcisme bij ALS en FTD identificeert focale mutaties die samenhangen met wijdverspreide degeneratie
Verborgen fouten in hersencellen
Amyotrofe laterale sclerose (ALS) en frontotemporale dementie (FTD) zijn verwoestende aandoeningen die mensen hun beweging, spraak en persoonlijkheid ontnemen. De meeste patiënten hebben geen familiegeschiedenis, wat een raadsel laat waarom hun zenuwcellen op een gegeven moment falen. Deze studie onderzoekt of kleine, later optredende DNA-fouten die alleen in kleine plekjes van de hersenen en het ruggenmerg ontstaan, kunnen bijdragen aan de ziekte bij deze zogenoemde sporadische gevallen.
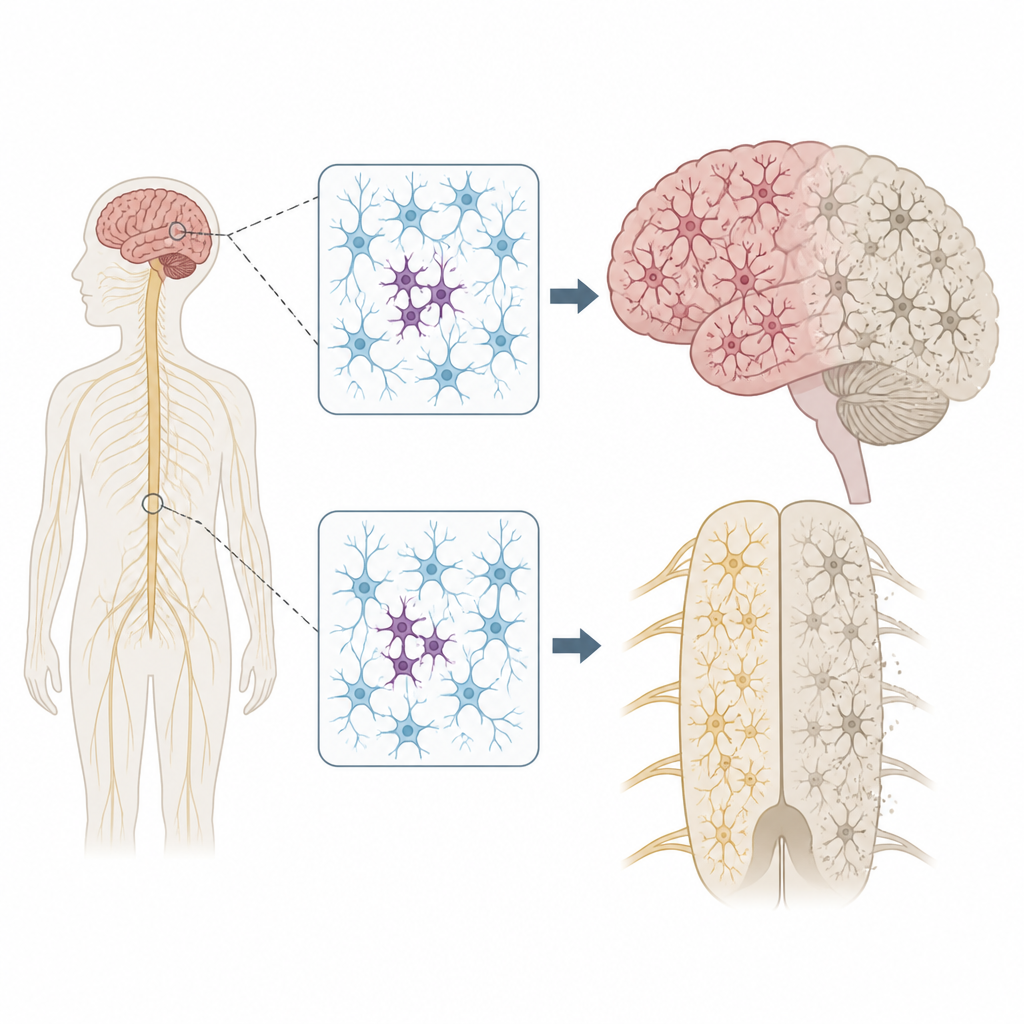
Waarom verspreide DNA-veranderingen ertoe doen
Ieder mens draagt een paar geërfde DNA-varianten die het ziekte‑risico kunnen verhogen of verlagen. Maar ons genoom blijft niet perfect stabiel. Naarmate cellen delen en verouderen, kunnen nieuwe mutaties optreden in slechts enkele cellen — een toestand die mosaïcisme wordt genoemd. De auteurs vroegen zich af of zulke plek‑gewijze mutaties in zenuwcellen kunnen helpen verklaren waarom ALS en FTD vaak in één lichaamsregio beginnen, zoals één hand of één kant van de tong, voordat ze zich breder door het zenuwstelsel verspreiden.
Belangrijke genen in het zenuwstelsel scannen
Om dit idee te testen onderzocht het team weefsel dat na overlijden werd gedoneerd van 291 mensen met ALS, 117 met FTD en 144 mensen zonder neurologische ziekte. Ze richtten zich op meerdere regio’s, waaronder de primaire motorische cortex, die vrijwillige bewegingen bestuurt, en het ruggenmerg, waar motorische neuronen zitten. Met een ultra-diepe sequentiemethode lazen ze 88 genen die aan neurodegeneratie zijn gekoppeld duizenden keren, waardoor ze zeldzame mutaties konden herkennen die in slechts een klein deel van de cellen in elk monster aanwezig zijn.
Geërfd risico en nieuwe mutaties
De eerste analyse zocht naar geërfde, in het hele lichaam aanwezige genveranderingen. Ongeveer 30 procent van de ALS- en FTD‑gevallen droeg bekende of sterk voorspelde schadelijke varianten in gevestigde ziektegenen, wat het belang van genetisch onderzoek benadrukt, zelfs als er geen familiegeschiedenis is. De onderzoekers haalden die gevallen uit verdere analyses om zich te concentreren op patiënten zonder zulk kiemlijnrisico. In deze overgebleven groep vonden ze dat ongeveer 2,1 procent nieuwe, schadelijke mutaties had die alleen in een subset van hersen‑ of ruggenmergcellen voorkwamen. Deze somatische varianten waren vrijwel afwezig in controlemateriaal.
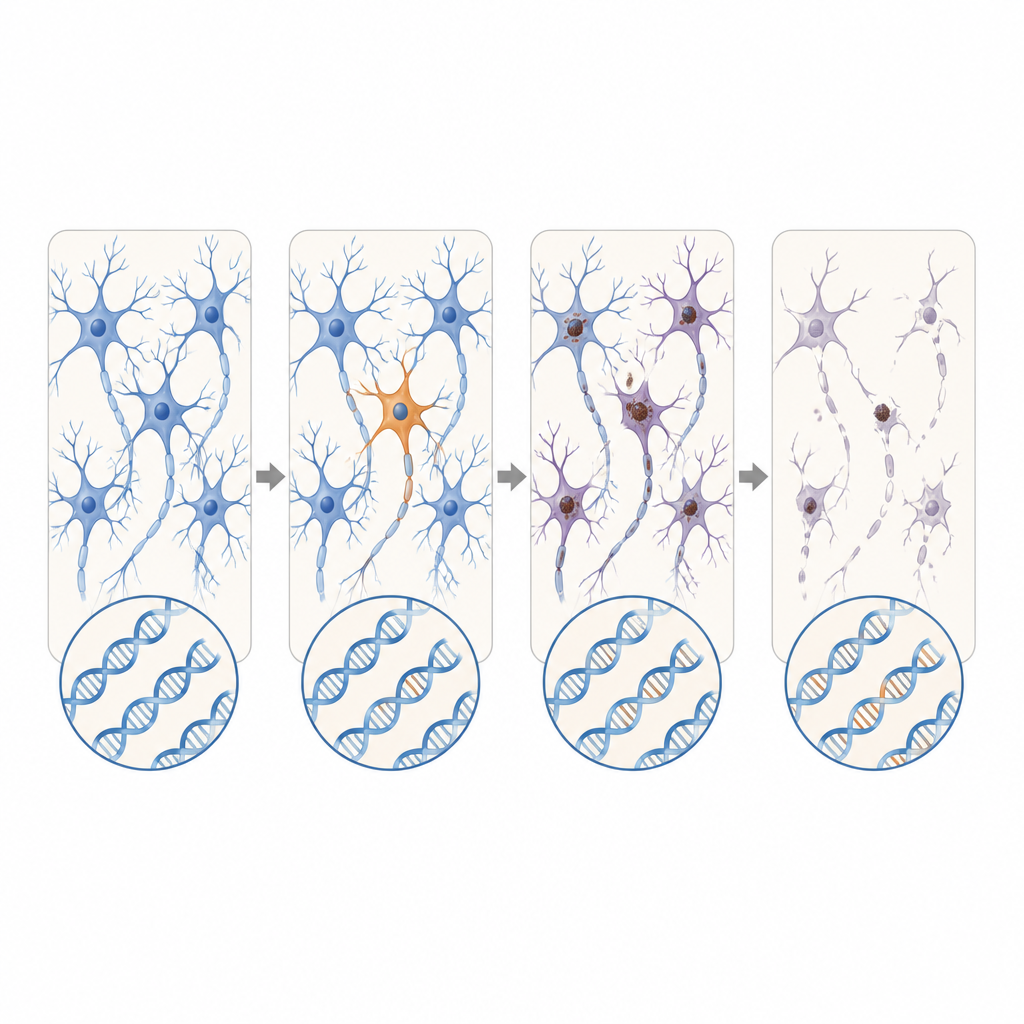
Focale problemen met verstrekkende gevolgen
De locatie van deze plek‑gewijze mutaties was opvallend. Bij ALS clusterden schadelijke varianten in ziektegenen in de primaire motorische cortex en het ruggenmerg — precies de regio’s met het grootste verlies aan zenuwcellen. Veel van deze mutaties waren aanwezig op extreem lage niveaus, vaak in minder dan 2 procent van de cellen, en waren beperkt tot één regio, wat suggereert dat ze laat in het leven zijn ontstaan. Toch vertoonde het omliggende weefsel wijdverspreide ophoping van een misgevouwen eiwit genaamd TDP-43 en sterk verlies van zenuwcellen, vergelijkbaar met gevallen met geërfde mutaties. Dit patroon ondersteunt het scenario waarin een kleine groep gemuteerde cellen de ziekte lokaal start, waarna toxische eiwitveranderingen zich via verbonden hersen- en ruggenmergcircuits naar buiten verspreiden.
Nieuwe kandidaatgenen en uitbreidende herhalingen
Buiten bekende ALS- en FTD-genen zochten de onderzoekers ook in de actieve genen van bulk-RNA‑gegevens van extra ALS-donoren. Ze ontdekten schadelijke somatische varianten in DYNC1H1 en LMNA — genen die in geërfde vorm ernstige kinderlijke motorneuronstoornissen veroorzaken en meestal overleving tot de leeftijden waarop ALS voorkomt verhinderen. Bij deze patiënten leken de foutieve versies slechts in een deel van de zenuwstelselcellen voor te komen, wat mogelijk normale ontwikkeling gevolgd door latere degeneratie toeliet. Met behulp van langread‑sequencing identificeerde het team ook een FTD‑geval waarin een kort, ogenschijnlijk onschuldig stukje herhaald DNA in het C9orf72-gen alleen in hersencellen dramatisch was uitgebreid, en zo een bereik bereikte dat bekend staat ziekte te veroorzaken.
Wat dit betekent voor patiënten en families
Samen suggereren de bevindingen dat, voor een klein maar betekenisvol aandeel van mensen met ogenschijnlijk sporadische ALS of FTD, de trigger zeldzame DNA‑veranderingen kan zijn die slechts in een klein stukje van het zenuwstelsel ontstaan in plaats van in elke lichaamscel. Deze verborgen fouten zijn te lokaal en te subtiel om in standaardbloedtests te verschijnen, maar kunnen toch voldoende zijn om een ketenreactie van eiwitmisvouwing en zenuwcelsterfte te starten die uiteindelijk grote delen van hersenen en ruggenmerg aantast. Hoewel huidige methoden vermoedelijk slechts een fractie van zulke gebeurtenissen detecteren, wijst dit werk op de behoefte aan gevoeliger instrumenten om mosaïsche mutaties te vinden en in kaart te brengen hoe pathologie zich vanuit deze kleine startpunten verspreidt.
Bronvermelding: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Trefwoorden: amyotrofe laterale sclerose, frontotemporale dementie, somatisch mosaïcisme, neurodegeneratie, C9orf72-herhalingsuitbreiding