Clear Sky Science · de
Somatische Mosaikbildung bei ALS und FTD identifiziert fokale Mutationen, die mit weitreichender Degeneration verbunden sind
Verborgene Fehler in Gehirnzellen
Amyotrophe Lateralsklerose (ALS) und frontotemporale Demenz (FTD) sind verheerende Erkrankungen, die Menschen Bewegungsfähigkeit, Sprache und Persönlichkeit rauben. Die meisten Patienten haben keine Familiengeschichte, sodass unklar bleibt, was ihre Nervenzellen zuerst auf den Weg des Versagens bringt. Diese Studie prüft, ob winzige, spät auftretende DNA-Fehler, die nur in kleinen Herden von Gehirn- und Rückenmarkszellen entstehen, zur Auslösung der Krankheit in diesen sogenannten sporadischen Fällen beitragen könnten.
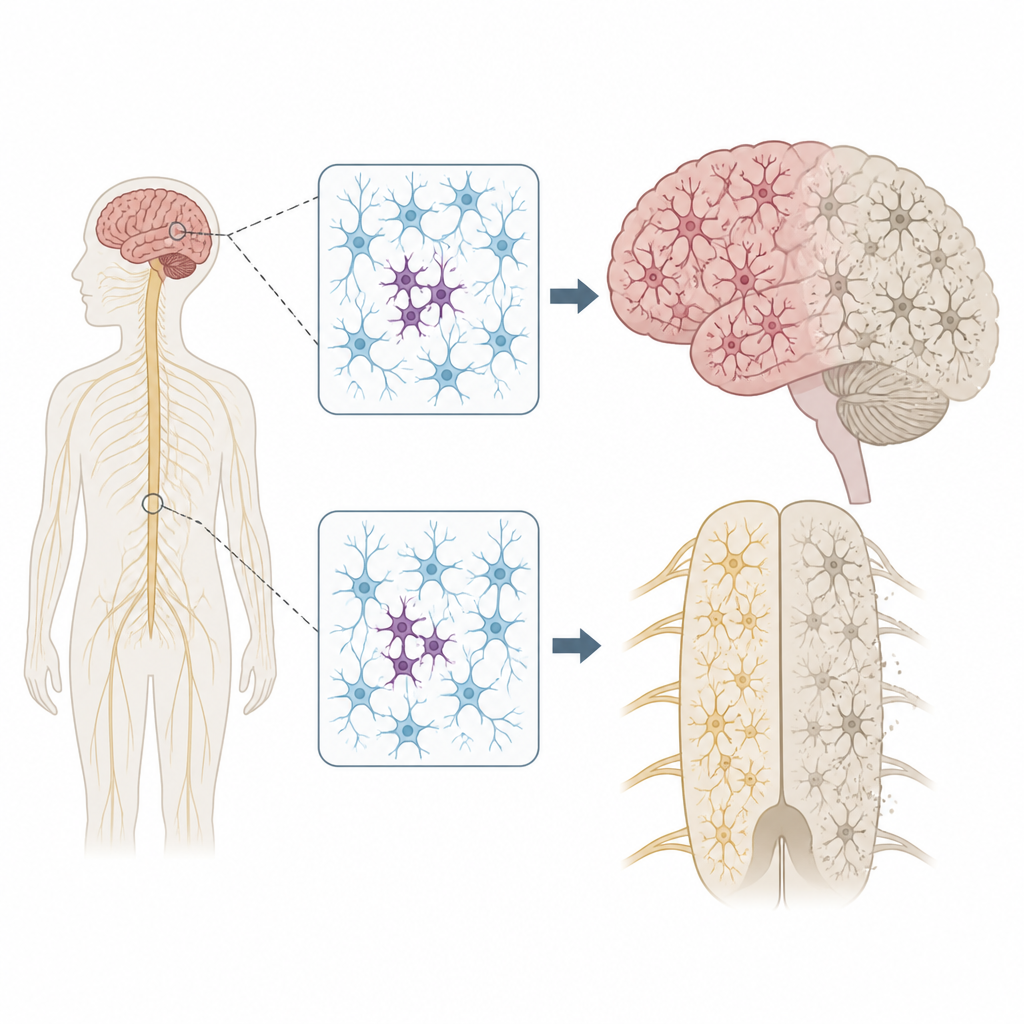
Warum verstreute DNA-Veränderungen wichtig sind
Jeder Mensch trägt einige vererbte DNA-Varianten, die das Erkrankungsrisiko erhöhen oder senken können. Unsere Genome bleiben jedoch nicht perfekt starr. Wenn Zellen sich teilen und altern, können neue Mutationen nur in bestimmten Zellen auftauchen — ein Zustand, der als Mosaikbildung bezeichnet wird. Die Autoren fragten sich, ob solche fadenförmig verteilten Mutationen in Nervenzellen erklären könnten, warum ALS und FTD oft in einer Körperregion beginnen, etwa in einer Hand oder auf einer Seite der Zunge, bevor sie sich weiter im Nervensystem ausbreiten.
Untersuchung wichtiger Gene im Nervensystem
Um diese Idee zu testen, analysierte das Team Gewebe, das nach dem Tod von 291 Menschen mit ALS, 117 mit FTD und 144 Personen ohne neurologische Erkrankung gespendet wurde. Sie konzentrierten sich auf mehrere Regionen, darunter den primären Motorcortex, der die willkürliche Bewegung steuert, und das Rückenmark, das Motoneurone enthält. Mithilfe eines ultratiefen Sequenzierverfahrens lasen sie 88 mit Neurodegeneration verknüpfte Gene tausendfach, was ihnen erlaubte, seltene Mutationen zu entdecken, die nur in einem kleinen Bruchteil der Zellen jeder Probe vorlagen.
Vererbtes Risiko und neue Mutationen
Der erste Durchlauf durch die Daten suchte nach vererbten, körperweit vorhandenen Genveränderungen. Etwa 30 Prozent der ALS- und FTD-Fälle trugen bekannte oder stark prognostizierte schädliche Varianten in etablierten Krankheitsgenen, was den Wert genetischer Tests selbst ohne Familienanamnese unterstreicht. Die Forscher schlossen diese Fälle dann aus der weiteren Analyse aus, um sich auf Patienten ohne solche Keimbahnrisiken zu konzentrieren. In dieser Restgruppe fanden sie, dass rund 2,1 Prozent neue, schädliche Mutationen aufwiesen, die nur in einem Teil der Gehirn- oder Rückenmarkszellen vorhanden waren. Diese somatischen Varianten fehlten in den Kontrollproben praktisch vollständig.
Fokale Probleme mit weitreichenden Folgen
Die Lokalisation dieser patchartigen Mutationen war auffällig. Bei ALS konzentrierten sich schädigende Varianten in Krankheitsgenen auf den primären Motorcortex und das Rückenmark — genau die Regionen, die den stärksten Nervenzellverlust zeigen. Viele dieser Mutationen lagen auf extrem niedrigem Niveau, oft in weniger als 2 Prozent der Zellen, und beschränkten sich auf eine Region, was darauf hindeutet, dass sie spät im Leben entstanden sind. Dennoch zeigte das umgebende Gewebe eine großflächige Anreicherung eines fehlgefalteten Proteins namens TDP-43 und einen starken Nervenzellverlust, ähnlich wie bei Fällen mit vererbten Mutationen. Dieses Muster stützt das Szenario, in dem eine kleine Gruppe mutierter Zellen die Krankheit lokal initiiert und toxische Proteinveränderungen sich dann durch verbundene Gehirn- und Rückenmarksschaltkreise ausbreiten.
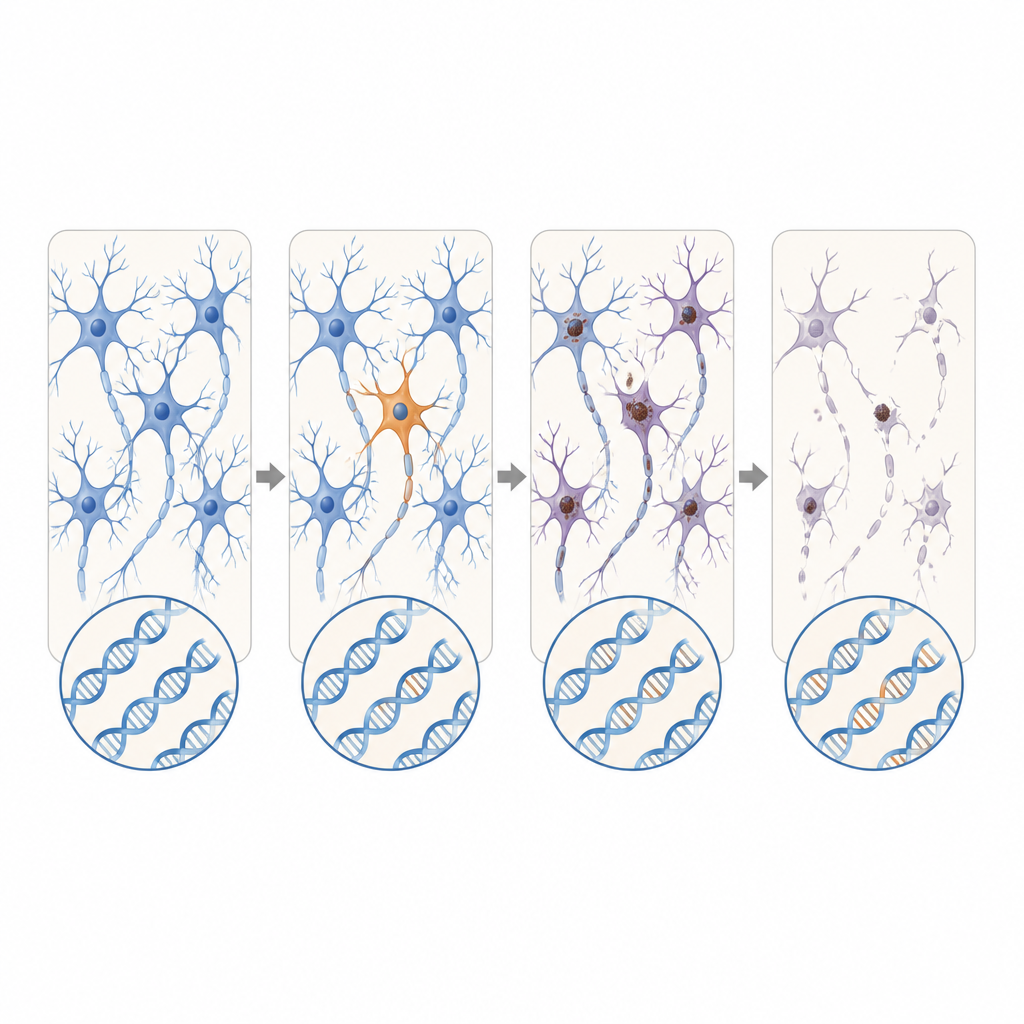
Neue Kandidatengene und expandierende Repeats
Über bekannte ALS- und FTD-Gene hinaus suchten die Wissenschaftler auch in aktiven Genen anhand von Bulk-RNA-Daten zusätzlicher ALS-Spender. Sie entdeckten schädigende somatische Varianten in DYNC1H1 und LMNA — Gene, die in ihrer vererbten Form schwere motorische Kinderkrankheiten verursachen, die üblicherweise ein Überleben bis ins ALS-Alter verhindern. Bei diesen Patienten traten die fehlerhaften Varianten jedoch nur in einem Teil der Nervensystemzellen auf, was möglicherweise eine normale Entwicklung gefolgt von einer spät einsetzenden Degeneration erlaubte. Mittels Long-Read-Sequenzierung identifizierte das Team außerdem einen FTD-Fall, in dem ein kurz erscheinender, zunächst harmloser Abschnitt repetitiver DNA im C9orf72-Gen nur in Gehirnzellen dramatisch expandierte und in einen Bereich vordrang, der bekanntermaßen Krankheit verursacht.
Was das für Patienten und Familien bedeutet
In der Summe deuten die Ergebnisse darauf hin, dass bei einem kleinen, aber bedeutsamen Anteil von offenbar sporadischen ALS- oder FTD-Fällen der Auslöser seltene DNA-Veränderungen sein kann, die nur in einem schmalen Abschnitt des Nervensystems entstehen und nicht in allen Körperzellen. Diese verborgenen Fehler sind zu lokal und zu subtil, um in Standard-Bluttests sichtbar zu werden, können aber dennoch ausreichen, eine Kaskade von Proteinfehlfaltung und Nervenzellverlust zu starten, die schließlich große Bereiche von Gehirn und Rückenmark betrifft. Obwohl gegenwärtige Methoden vermutlich nur einen Teil solcher Ereignisse erfassen können, verweist diese Arbeit auf die Notwendigkeit sensitiverer Werkzeuge, um mosaikartige Mutationen zu finden und nachzuzeichnen, wie sich die Pathologie von diesen winzigen Ausgangspunkten aus ausbreitet.
Zitation: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Schlüsselwörter: amyotrophe Lateralsklerose, frontotemporale Demenz, somatische Mosaikbildung, Neurodegeneration, C9orf72-Repeat-Expansion