Clear Sky Science · pl
Możaika somatyczna w ALS i FTD identyfikuje ogniskowe mutacje powiązane z rozległym zanikiem
Ukryte usterki w komórkach mózgu
Stwardnienie zanikowe boczne (ALS) i otępienie czołowo‑skroniowe (FTD) to wyniszczające choroby, które odbierają ludziom zdolność poruszania się, mówienia i cechy osobowości. U większości pacjentów nie występuje historia rodzinna, co pozostawia zagadkę, co pierwotnie skierowuje ich komórki nerwowe na drogę ku upadkowi. Badanie to stawia pytanie, czy drobne, pojawiające się późno błędy w DNA, które występują jedynie w niewielkich płatach mózgu i rdzenia kręgowego, mogą przyczyniać się do zapoczątkowania choroby w tych tak zwanych sporadycznych przypadkach.
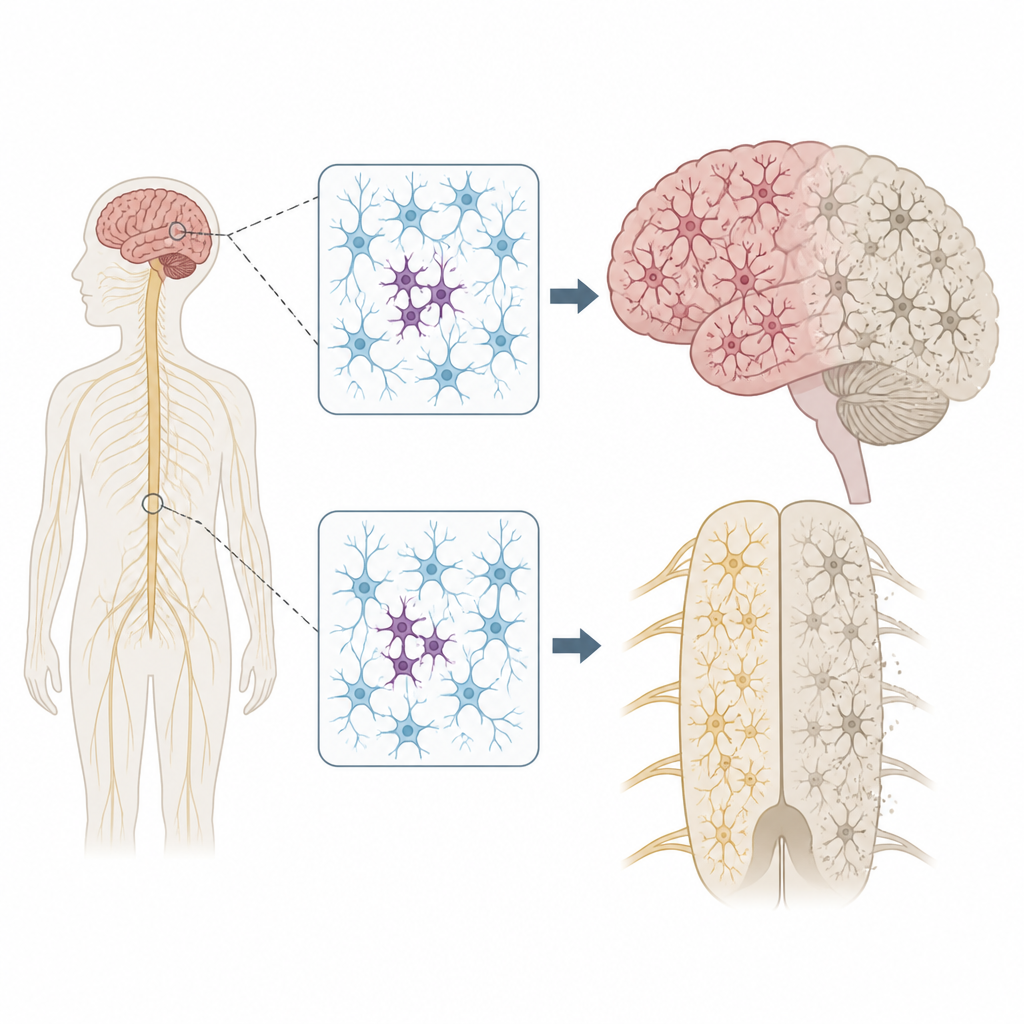
Dlaczego rozrzucone zmiany DNA mają znaczenie
Każdy nosi kilka dziedzicznych wariantów DNA, które mogą zwiększać lub zmniejszać ryzyko choroby. Jednak nasze genomy nie pozostają całkowicie niezmienne. W miarę podziałów i starzenia się komórek w niektórych z nich mogą pojawiać się nowe mutacje — stan zwany mozaicyzmem. Autorzy zastanawiali się, czy takie plamiste mutacje w komórkach nerwowych mogłyby pomóc wyjaśnić, dlaczego ALS i FTD często zaczynają się w jednym obszarze ciała, na przykład w jednej dłoni lub po jednej stronie języka, zanim rozszerzą się szerzej po układzie nerwowym.
Skanowanie kluczowych genów układu nerwowego
Aby sprawdzić tę hipotezę, zespół przeanalizował tkanki przekazane po śmierci od 291 osób z ALS, 117 z FTD i 144 osób bez chorób neurologicznych. Skupili się na wielu regionach, w tym na pierwotnej korze ruchowej, która kontroluje ruchy dowolne, oraz na rdzeniu kręgowym, gdzie znajdują się neurony ruchowe. Przy użyciu ultra‑głębokiego sekwencjonowania odczytali 88 genów powiązanych z neurodegeneracją tysiące razy, co pozwoliło im wykryć rzadkie mutacje obecne jedynie w niewielkiej frakcji komórek w każdej próbce.
Ryzyko dziedziczne i nowe mutacje
Pierwsze przeszukanie danych skupiało się na dziedzicznych, obecnych w całym organizmie zmianach genów. Około 30 procent przypadków ALS i FTD miało znane lub silnie przewidywane szkodliwe warianty w ustalonych genach chorobowych, co potwierdza wartość testów genetycznych nawet gdy brak jest historii rodzinnej. Badacze następnie wyłączyli te przypadki z dalszej analizy, aby skupić się na pacjentach bez takiego germinalnego ryzyka. W tej pozostałej grupie stwierdzili, że około 2,1 procent miało nowe, szkodliwe mutacje pojawiające się jedynie w podzbiorze komórek mózgu lub rdzenia. Te warianty somatyczne były niemal całkowicie nieobecne w tkankach kontrolnych.
Ogniskowe problemy o dalekosiężnych skutkach
Lokalizacja tych plamistych mutacji była znamienna. W ALS szkodliwe warianty w genach chorobowych skupiały się w pierwotnej korze ruchowej i rdzeniu kręgowym — właśnie w tych regionach, które wykazują największe ubytki neuronów. Wiele z tych mutacji występowało na niezwykle niskim poziomie, często w mniej niż 2 procentach komórek, i ograniczało się do jednego regionu, co sugeruje, że pojawiły się późno w życiu. Mimo to otaczająca tkanka wykazywała rozległe gromadzenie się zmisfoldowanego białka TDP‑43 i znaczną utratę neuronów, podobnie jak w przypadkach z mutacjami dziedzicznymi. Ten wzorzec wspiera scenariusz, w którym mała grupa zmutowanych komórek lokalnie inicjuje chorobę, a toksyczne zmiany białkowe następnie rozprzestrzeniają się przez połączenia w mózgu i rdzeniu kręgowym.
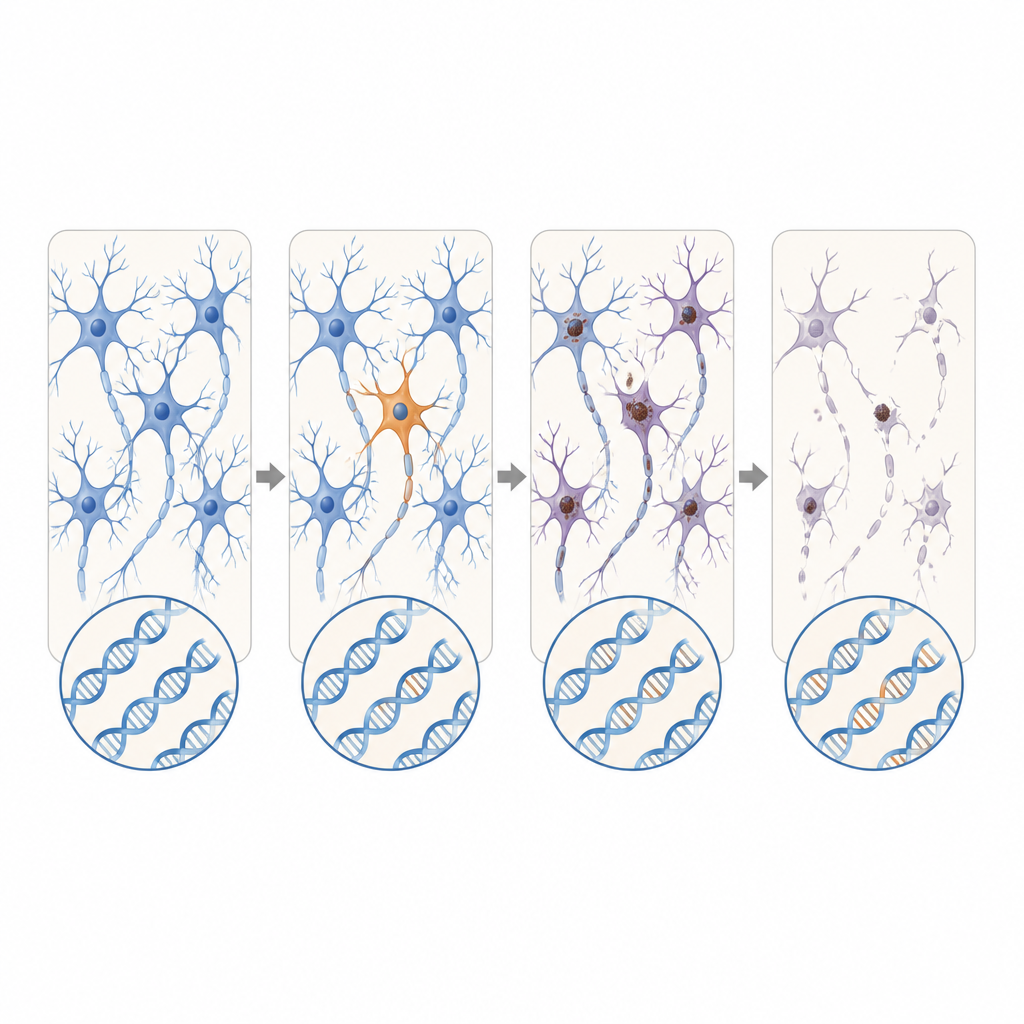
Nowe geny kandydujące i rozszerzające się powtórzenia
Ponad znane geny ALS i FTD, naukowcy przeszukali również aktywne geny w danych RNA z dodatkowych dawców z ALS. Odkryli szkodliwe warianty somatyczne w DYNC1H1 i LMNA — genach, które w formie dziedzicznej powodują ciężkie dziecięce choroby neuronów ruchowych zwykle uniemożliwiające przeżycie do wieku, w którym występuje ALS. U tych pacjentów wadliwe wersje pojawiły się tylko w części komórek układu nerwowego, co potencjalnie pozwoliło na normalny rozwój, a następnie późne wystąpienie degeneracji. Przy użyciu sekwencjonowania długich odczytów zespół zidentyfikował także przypadek FTD, w którym krótki, pozornie niegroźny odcinek powtórzeń w genie C9orf72 rozszerzył się dramatycznie jedynie w komórkach mózgowych, przekraczając zakres znany z wywoływania choroby.
Co to oznacza dla pacjentów i rodzin
Podsumowując, wyniki sugerują, że u niewielkiej, ale istotnej części osób z pozornie sporadycznym ALS lub FTD czynnikiem wyzwalającym mogą być rzadkie zmiany DNA powstające tylko w wąskim wycinku układu nerwowego, a nie w każdej komórce organizmu. Te ukryte usterki są zbyt lokalne i zbyt subtelne, by wykryć je standardowymi badaniami krwi, a jednak mogą wystarczyć, by zapoczątkować łańcuchową reakcję zmisfoldowania białek i śmierci neuronów, która ostatecznie obejmuje duże regiony mózgu i rdzenia. Choć obecne metody prawdopodobnie wykrywają tylko część takich zdarzeń, praca ta wskazuje na potrzebę bardziej czułych narzędzi do wykrywania mutacji mozaikowych i mapowania, jak patologia rozprzestrzenia się z tych drobnych punktów wyjścia.
Cytowanie: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Słowa kluczowe: stwardnienie zanikowe boczne, otępienie czołowo‑skroniowe, mozaicyzm somatyczny, neurodegeneracja, ekspansja powtórzeń C9orf72