Clear Sky Science · pt
Mosaicismo somático em ALS e FTD identifica mutações focais associadas à degeneração disseminada
Falhas ocultas nas células cerebrais
A esclerose lateral amiotrófica (ALS) e a demência frontotemporal (FTD) são condições devastadoras que privam as pessoas de movimento, fala e personalidade. A maioria dos pacientes não tem histórico familiar, deixando em aberto o mistério sobre o que inicialmente coloca suas células nervosas em rota de falha. Este estudo investiga se pequenas falhas no DNA que surgem tardiamente e apenas em pequenos pontos do cérebro e da medula espinhal podem ajudar a desencadear a doença nesses casos chamados esporádicos.
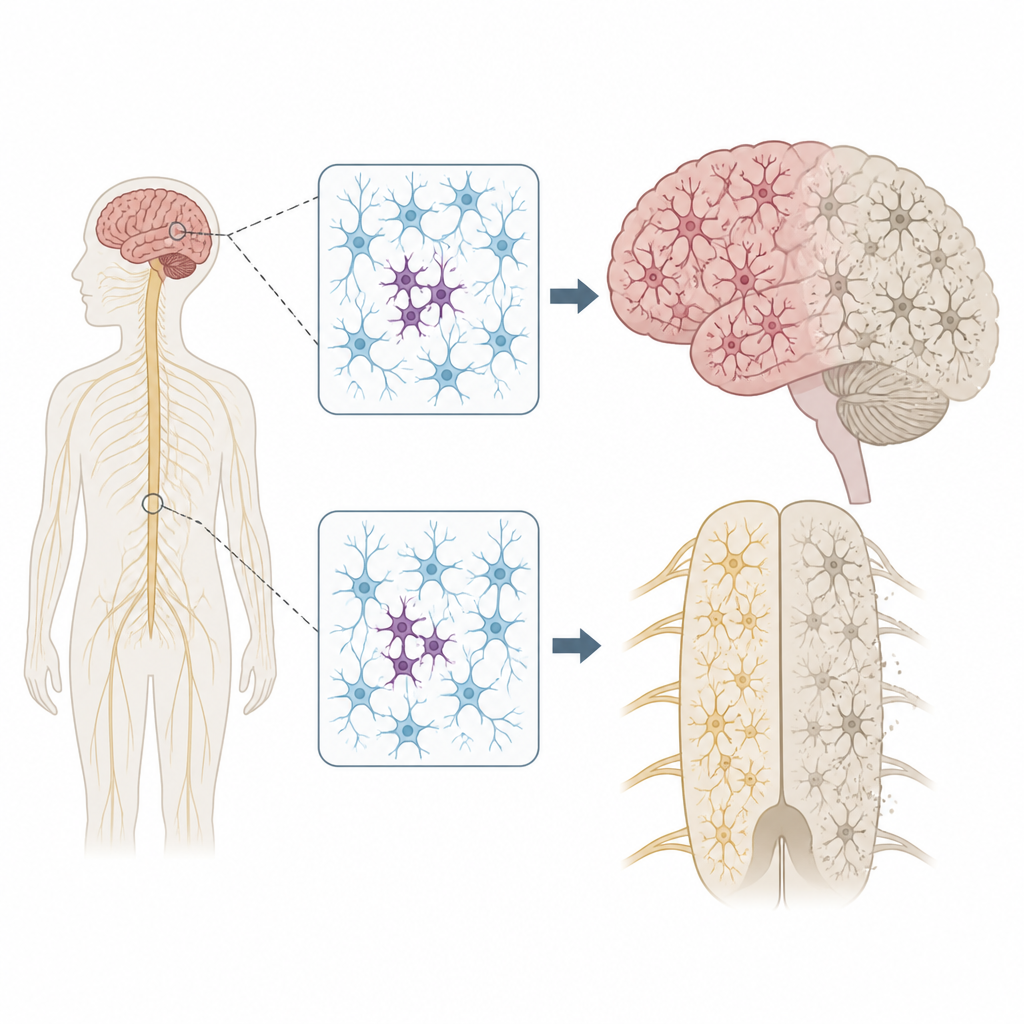
Por que mudanças dispersas no DNA importam
Cada pessoa carrega algumas variantes herdadas no DNA que podem aumentar ou reduzir o risco de doença. Mas nossos genomas não permanecem perfeitamente fixos. À medida que as células se dividem e envelhecem, novas mutações podem aparecer apenas em algumas células, um estado chamado mosaicismo. Os autores se perguntaram se tais mutações pontuais em células nervosas poderiam ajudar a explicar por que ALS e FTD frequentemente começam em uma região do corpo, como uma única mão ou um lado da língua, antes de se espalharem mais amplamente pelo sistema nervoso.
Escaneando genes-chave no sistema nervoso
Para testar essa ideia, a equipe examinou tecidos doados após a morte de 291 pessoas com ALS, 117 com FTD e 144 indivíduos sem doença neurológica. Eles focaram em múltiplas regiões, incluindo o córtex motor primário, que controla o movimento voluntário, e a medula espinhal, que abriga os neurônios motores. Usando um método de sequenciamento ultra-profundo, leram 88 genes ligados à neurodegeneração milhares de vezes, o que lhes permitiu detectar mutações raras presentes apenas em uma pequena fração de células em cada amostra.
Risco herdado e novas mutações
A primeira análise dos dados buscou mudanças genéticas herdadas e presentes em todo o corpo. Aproximadamente 30% dos casos de ALS e FTD carregavam variantes conhecidas ou fortemente previstas como danosas em genes estabelecidos da doença, reforçando o valor dos testes genéticos mesmo quando não há história familiar. Os pesquisadores então excluíram esses casos das análises subsequentes para se concentrar em pacientes que não apresentavam tal risco germinativo. Nesse grupo restante, encontraram cerca de 2,1% que abrigavam novas mutações danosas que apareciam apenas em um subconjunto de células do cérebro ou da medula espinhal. Essas variantes somáticas estavam quase totalmente ausentes nos tecidos de controle.
Problemas focais com efeitos de longo alcance
A localização dessas mutações pontuais foi marcante. Na ALS, variantes danosas em genes da doença se concentraram no córtex motor primário e na medula espinhal, as regiões que apresentam maior perda de células nervosas. Muitas dessas mutações estavam presentes em níveis extremamente baixos, frequentemente em menos de 2% das células, e confinadas a uma região, sugerindo que surgiram tardiamente na vida. Ainda assim, o tecido circundante mostrou acúmulo disseminado de uma proteína mal dobrada chamada TDP-43 e intensa perda neuronal, muito semelhante a casos com mutações herdadas. Esse padrão apoia um cenário em que um pequeno grupo de células mutantes inicia a doença localmente, e alterações proteicas tóxicas então se propagam por circuitos conectados do cérebro e da medula espinhal.
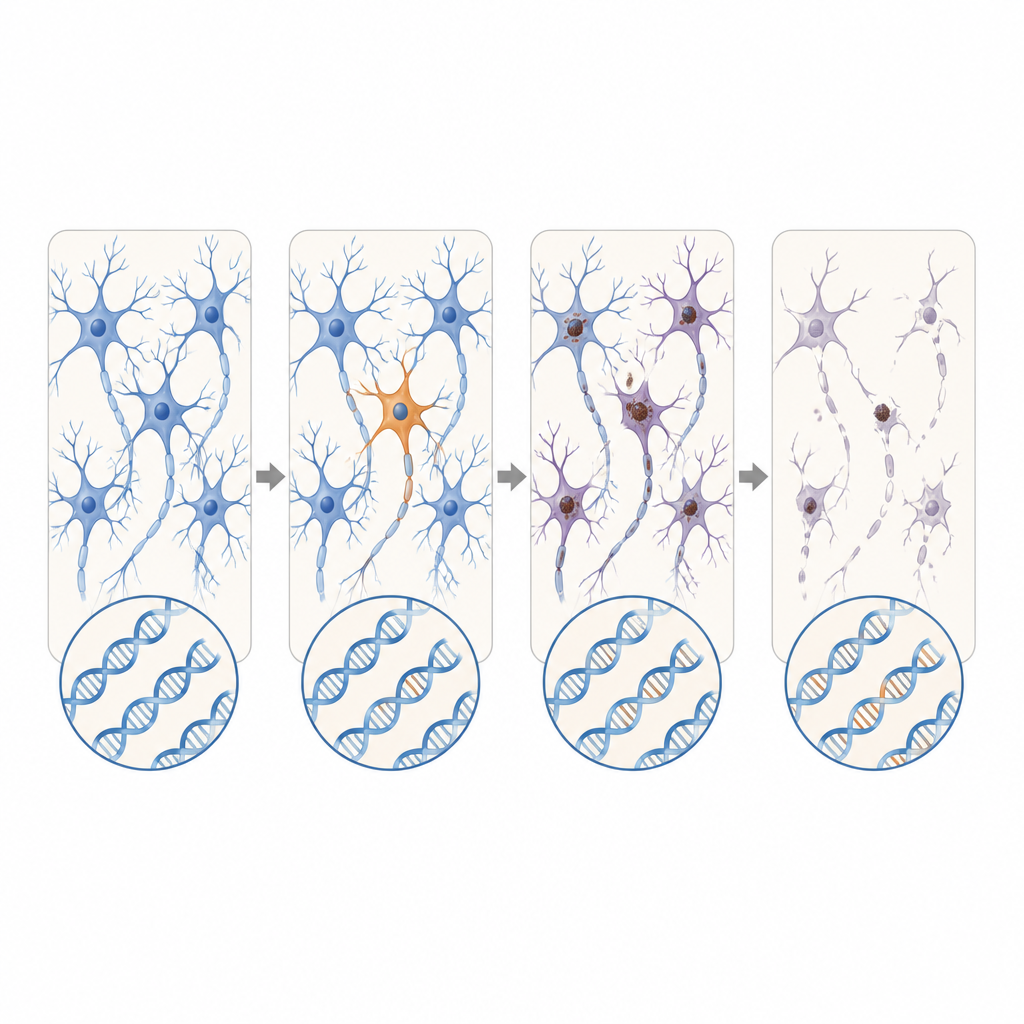
Novos genes candidatos e repetições em expansão
Além dos genes conhecidos de ALS e FTD, os cientistas também buscaram por todo o conjunto de genes ativos em dados de RNA em massa de doadores adicionais com ALS. Eles descobriram variantes somáticas danosas em DYNC1H1 e LMNA, genes que em sua forma herdada causam distúrbios motores infantis graves que geralmente impedem a sobrevivência até a idade em que a ALS ocorre. Nesses pacientes, as versões defeituosas apareceram apenas em uma porção das células do sistema nervoso, potencialmente permitindo o desenvolvimento normal seguido por degeneração de início tardio. Usando sequenciamento de leituras longas, a equipe também identificou um caso de FTD no qual um trecho curto, aparentemente inofensivo, de DNA repetido no gene C9orf72 expandiu-se dramaticamente apenas em células cerebrais, alcançando uma faixa conhecida por causar doença.
O que isso significa para pacientes e famílias
Em conjunto, os achados sugerem que, para uma parcela pequena mas relevante de pessoas com ALS ou FTD aparentemente esporádicos, o gatilho pode ser mudanças raras no DNA que surgem em apenas uma fatia do sistema nervoso em vez de em todas as células do corpo. Essas falhas ocultas são locais e sutis demais para aparecer em exames sanguíneos padrão, ainda que possam ser suficientes para iniciar uma reação em cadeia de dobramento proteico anômalo e morte neuronal que eventualmente afeta grandes regiões do cérebro e da medula espinhal. Embora os métodos atuais provavelmente detectem apenas uma fração desses eventos, este trabalho aponta para a necessidade de ferramentas mais sensíveis para encontrar mutações mosaicas e mapear como a patologia se espalha a partir desses pequenos pontos de partida.
Citação: Zhou, Z., Kim, J., Huang, A.Y. et al. Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration. Nat Genet 58, 1019–1029 (2026). https://doi.org/10.1038/s41588-026-02570-6
Palavras-chave: esclerose lateral amiotrófica, demência frontotemporal, mosaicismo somático, neurodegeneração, expansão de repetição em C9orf72