Clear Sky Science · sv
Att komprimera den kollektiva kunskapen från ESM till en enda proteinspråkmodell
Varför det spelar roll att slå ihop många proteinmodeller till en
Varje människa bär på miljontals små skillnader i sitt DNA, många av dem som ändrar enstaka byggstenar i proteiner. De flesta av dessa förändringar är ofarliga, men några kan leda till sjukdom. Läkare och forskare skulle vilja ha ett snabbt och tillförlitligt sätt att avgöra vilka som är skadliga, enbart utifrån proteinsekvensen. Den här artikeln beskriver en ny strategi som tar den samlade erfarenheten från många befintliga proteinspråkmodeller och komprimerar den till ett enda, effektivt system som matchar eller överträffar dagens bästa verktyg för att bedöma effekten av genetiska förändringar.
Från att läsa meningar till att läsa proteiner
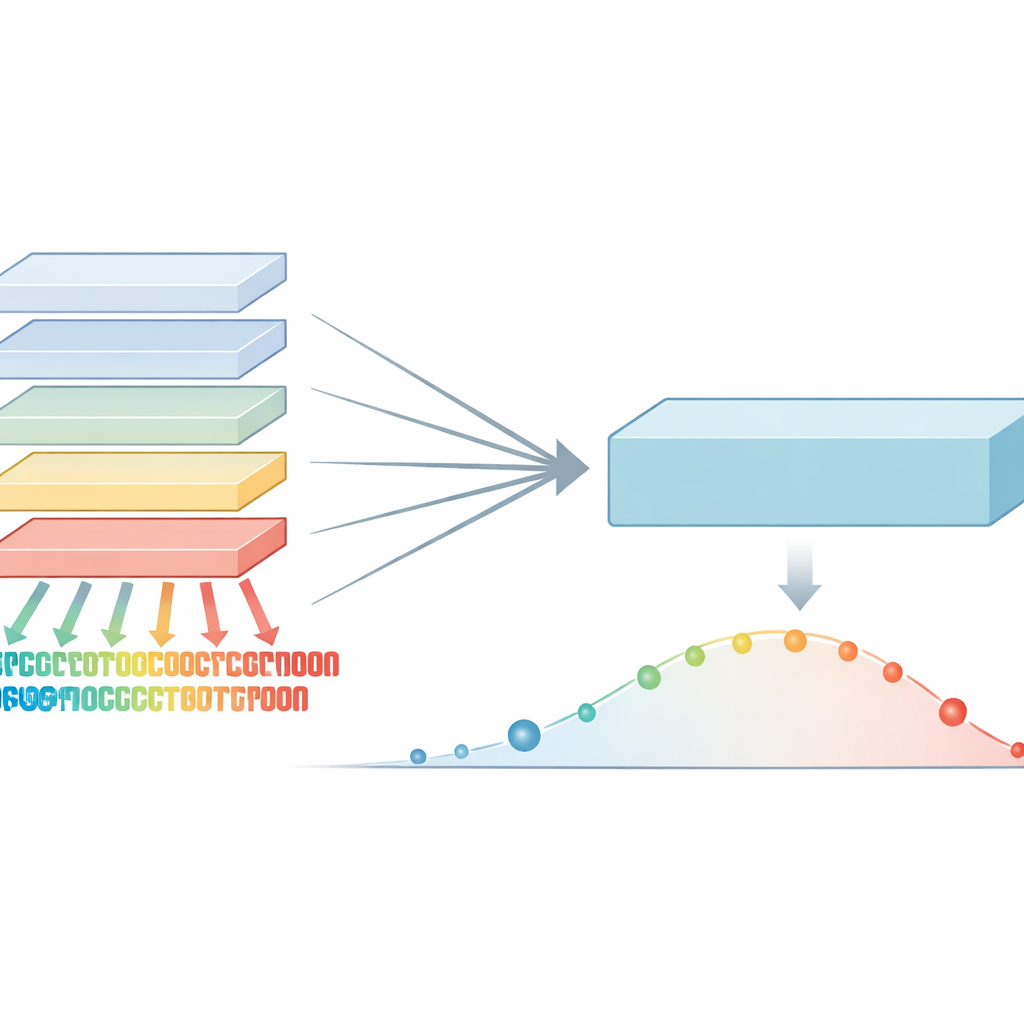
Proteinspråkmodeller lånar idéer från tekniken bakom moderna översättnings‑ och chattssystem. Istället för att lära sig ord i en mening lär de mönster i strängar av aminosyror, bokstäverna i proteinsekvenser. Genom att träna på hundratals miljoner naturliga proteiner plockar dessa modeller upp signaler om vilka positioner som är starkt bevarade och vilka som tål förändring. De mönstren kan sedan användas för att poängsätta genetiska varianter: om en förändring bryter ett mönster som evolutionen starkt skyddar, är den mer sannolik att vara skadlig. Fram till nu kombinerade de starkaste metoderna dessa modeller med extra information såsom 3D‑struktur eller evolutionära familjeträd, vilket gjorde dem kraftfulla men också komplexa och ibland svåra att använda brett.
Låta modeller lära av varandra
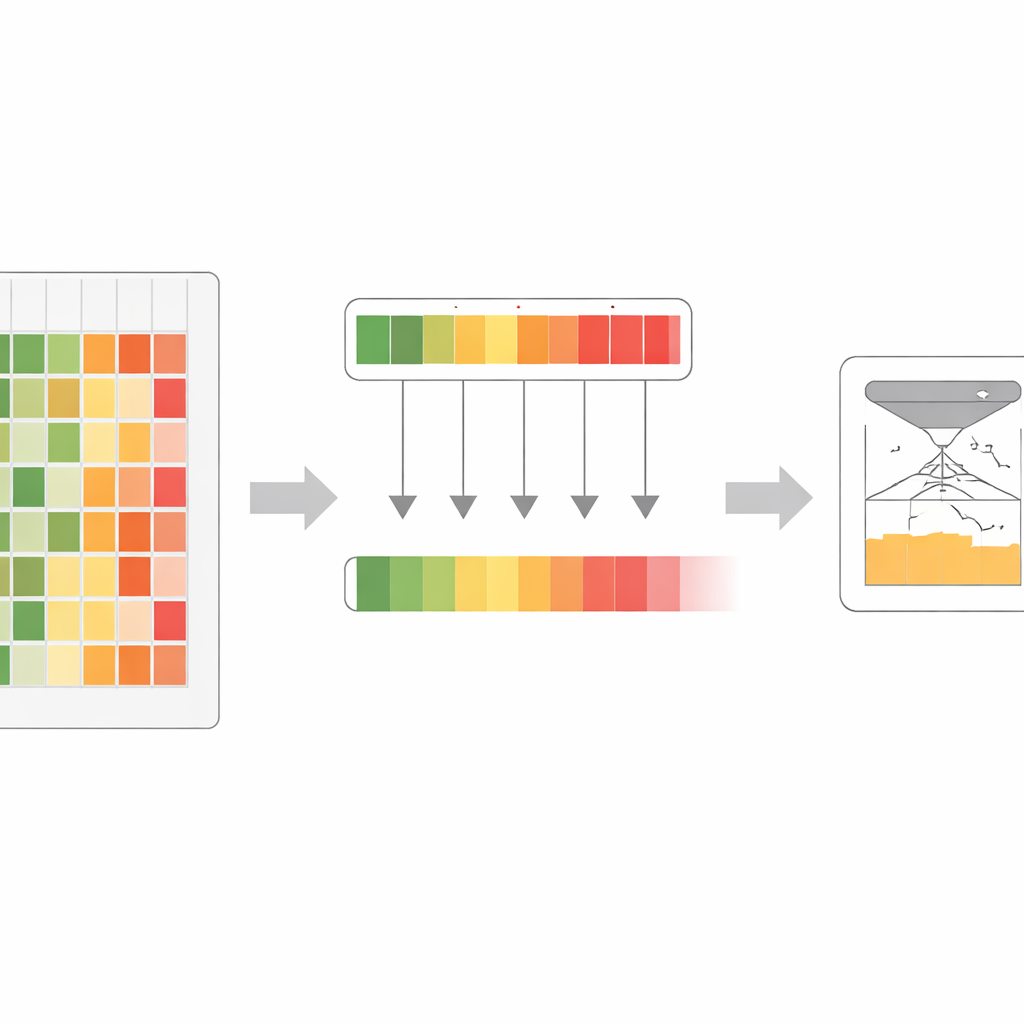
Författarna noterade att olika proteinspråkmodeller, även när de byggts på liknande data, har olika styrkor och blinda fläckar. En modell kan vara särskilt skicklig på att upptäcka känsliga regioner i en proteinfamilj, medan en annan utmärker sig någon annanstans. Istället för att medelvärdera deras åsikter introducerade teamet en "maximalt‑konfidens"‑regel: för varje möjlig proteinförändring ser de över hela familjen av modeller och behåller den som är mest säker på att förändringen är skadlig. Detta steg ger en berikad uppsättning poäng som fångar de starkaste evolutionära varningssignalerna från vilken modell som helst i gruppen, istället för att utjämna dem.
Koka ner många röster till en
Med denna berikade signal designade forskarna en träningsprocess de kallar co‑distillation. Här övar alla originalmodeller upprepade gånger på att matcha den starkast sammansatta signalen, och turas om att vara "student" och "lärare" beroende på var var och en är mest säker. I tidiga omgångar hjälper maximalt‑konfidens‑regeln till att framhäva subtila men viktiga mönster som vissa modeller missat. I senare omgångar hjälper ett mildare medelvärdessteg modellerna att komma överens och jämna ut brus. Genom flera cykler absorberar till slut en stor modell gradvis nästan all användbar information som tidigare spreds över hela ensemblet. Denna slutliga modell, kallad VESM‑3B, används sedan för att träna mindre versioner som körs tillräckligt snabbt för stora genetiska dataset samtidigt som de behåller större delen av noggrannheten.
Slår komplexa system med enbart sekvenser
Trots att de använder enbart råa proteinsekvenser som input matchar eller överträffar VESM‑modellerna sofistikerade konkurrenter som också förlitar sig på 3D‑strukturer, handbyggda evolutionära historiker eller data från mänskliga populationer. På kliniska mått dragna från ClinVar‑databasen över sjukdomsvarianter presterar huvudmodellen VESM bättre än många allmänt använda verktyg och slår till och med AlphaMissense, ett nyligt högprofilerat system som blandar struktur‑ och populationsdata. Avgörande är att VESM:s prestanda håller även för mycket sällsynta varianter — de som kliniker har svårast att tolka. Modellerna utmärker sig också på laboratoriemätningar som testar hur mutationer påverkar proteins funktion, stabilitet och bindning, och de kan spåra storleken och riktningen av varianteffekter på verkliga kliniska egenskaper i stora biobankdataset.
Vad detta betyder för genetik och medicin
Genom att noggrant kombinera och förfina styrkorna hos många sekvensmodeller visar detta arbete att råa proteinsekvenser ensamma kan bära tillräckligt med signal för att förutsäga effekten av genetiska förändringar på vetenskaplig toppnivå. Resultatet är en familj verktyg som är enklare att distribuera än struktur‑tunga eller datahungriga system, men som ändå förblir mycket precisa över proteiner från människor, mikrober och virus. För kliniker och forskare innebär detta snabbare, mer pålitlig prioritering av DNA‑varianter, en tydligare bild av hur starkt en förändring kan påverka sjukdomsrelaterade egenskaper, och ett praktiskt sätt att föra in kraftfulla prediktionsmodeller i vardagliga arbetsflöden för genetik och protein‑design.
Citering: Dinh, T., Jang, SK., Zaitlen, N. et al. Compressing the collective knowledge of ESM into a single protein language model. Nat Methods 23, 772–784 (2026). https://doi.org/10.1038/s41592-026-03050-9
Nyckelord: proteinspråkmodeller, prediktion av variantpåverkan, genetiska varianter, evolutionära signaler, klinisk genomik