Clear Sky Science · de
Das kollektive Wissen von ESM in ein einzelnes Protein-Sprachmodell komprimieren
Warum es wichtig ist, viele Proteinmodelle in eins zu überführen
Jeder Mensch trägt Millionen winziger Unterschiede in seiner DNA, von denen viele einzelne Bausteine in Proteinen verändern. Die meisten dieser Veränderungen sind harmlos, doch einige können Krankheiten verursachen. Ärztinnen, Ärzte und Forschende wünschen sich eine schnelle, zuverlässige Methode, um allein anhand der Proteinsequenz zu unterscheiden, welche Veränderungen gefährlich sind. Dieser Artikel beschreibt eine neue Strategie, die das gesammelte Fachwissen vieler existierender Protein-„Sprachmodelle“ in ein einzelnes, effizientes System komprimiert, das mit den derzeit besten Werkzeugen zur Beurteilung genetischer Veränderungen mithält oder sie übertrifft.
Vom Lesen von Sätzen zum Lesen von Proteinen
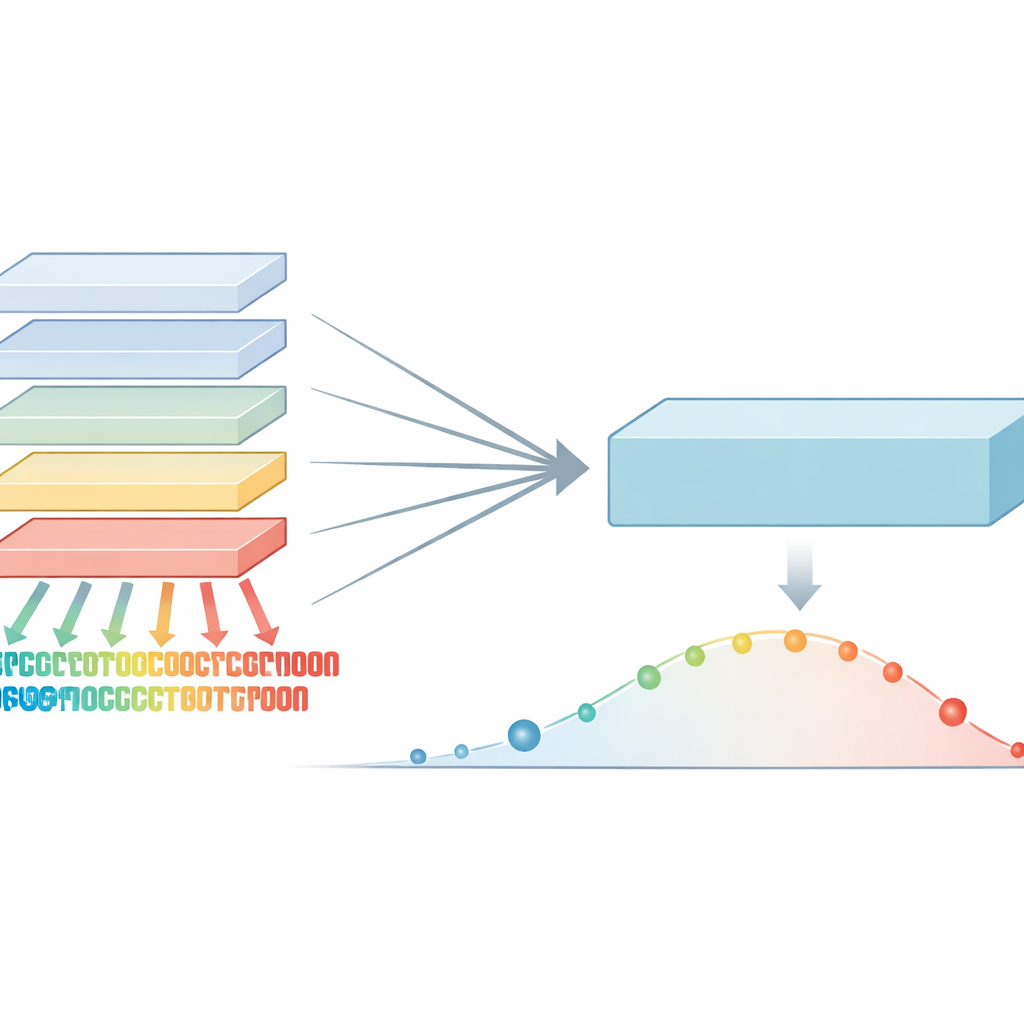
Protein-Sprachmodelle entlehnen Ideen aus der Technologie hinter modernen Übersetzungs- und Chat-Systemen. Statt Wörter in einem Satz zu lernen, erfassen sie Muster in Aminosäureketten, den „Buchstaben" von Proteinsequenzen. Durch Training an Hunderten Millionen natürlicher Proteine erkennen diese Modelle, welche Positionen stark konserviert sind und welche Veränderungen tolerieren. Diese Muster lassen sich dann nutzen, um genetische Varianten zu bewerten: Bricht eine Veränderung ein von der Evolution stark geschütztes Muster, ist sie wahrscheinlicher schädlich. Bislang kombinierten die stärksten Methoden diese Modelle oft mit zusätzlicher Information wie 3D-Strukturen oder evolutionären Stammbaumdaten, was sie leistungsfähig, aber auch komplex und nicht immer breit anwendbar machte.
Modelle voneinander lernen lassen
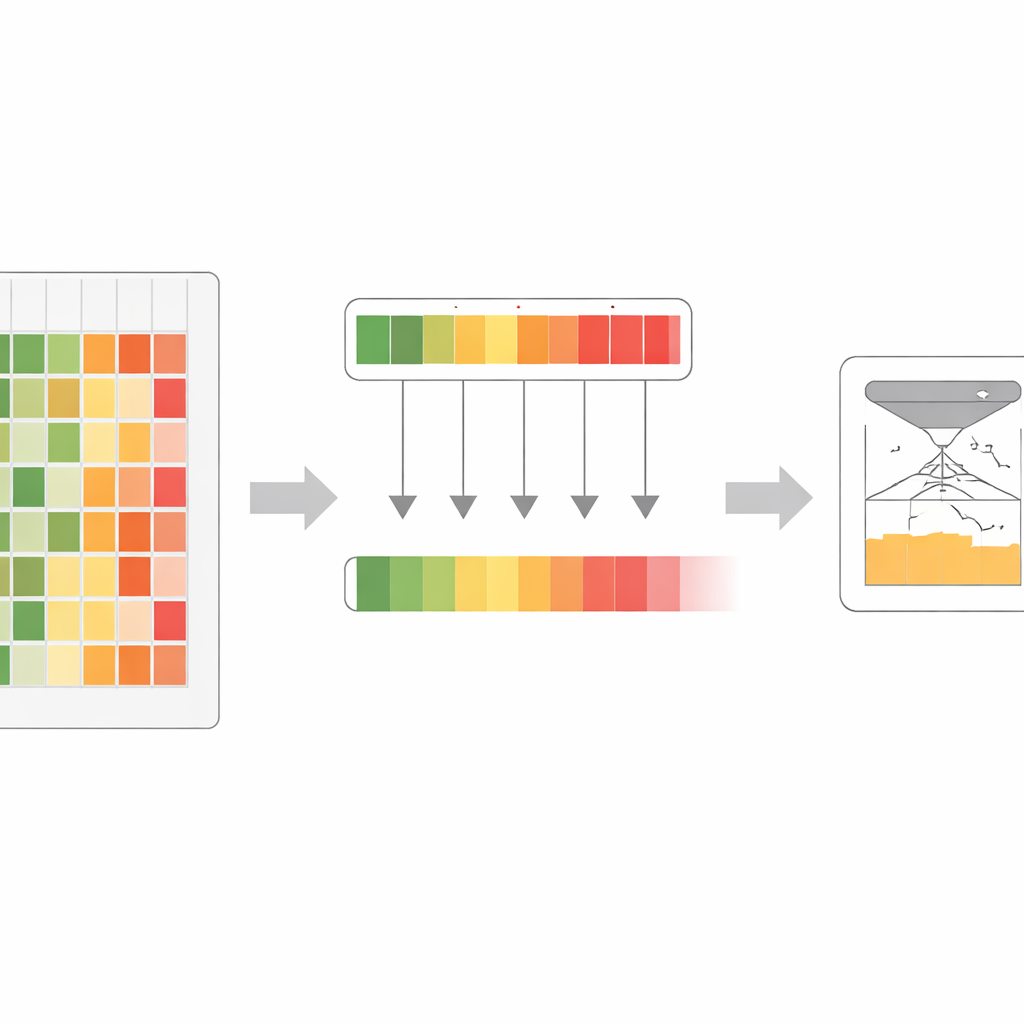
Die Autorinnen und Autoren stellten fest, dass verschiedene Protein-Sprachmodelle, selbst bei ähnlichen Trainingsdaten, unterschiedliche Stärken und blinde Flecken haben. Ein Modell ist vielleicht besonders gut darin, sensible Regionen in einer Proteinfamilie zu erkennen, während ein anderes anderswo brilliert. Anstatt ihre Meinungen einfach zu mitteln, führten sie eine „Maximalvertrauens“-Regel ein: Für jede mögliche Proteinveränderung betrachten sie eine ganze Familie von Modellen und behalten denjenigen Wert, bei dem das Modell am sichersten ist, dass die Veränderung schädlich ist. Dieser Schritt erzeugt ein angereichertes Satz von Scores, das die stärksten evolutionären Warnsignale aus jedem Modell der Gruppe einfängt, anstatt sie zu verwässern.
Viele Stimmen auf einen Punkt reduzieren
Ausgehend von diesem angereicherten Signal entwarfen die Forschenden einen Trainingsprozess, den sie Ko-Distillation nennen. Dabei üben alle ursprünglichen Modelle wiederholt, das stärkste kombinierte Signal nachzuahmen, wobei sie je nach Bereich als „Schüler“ oder „Lehrer“ fungieren, je nachdem, wo jedes Modell am sichersten ist. In frühen Runden hilft die Maximalvertrauens-Regel, subtile aber wichtige Muster hervorzuheben, die einige Modelle übersehen hatten. In späteren Runden führt ein sanfteres Mittelungsverfahren zu besserer Übereinstimmung und zur Glättung von Rauschen. Durch mehrere Zyklen nimmt ein großes Modell nach und nach fast alle nützlichen Informationen auf, die zuvor über das gesamte Ensemble verteilt waren. Dieses Endmodell, VESM-3B genannt, dient dann dazu, kleinere Versionen zu trainieren, die schnell genug für große genetische Datensätze laufen und dabei den Großteil der Genauigkeit bewahren.
Komplexe Systeme allein mit Sequenzen übertreffen
Obwohl die VESM-Modelle nur rohe Proteinsequenzen als Eingabe verwenden, erreichen oder übertreffen sie anspruchsvolle Konkurrenzsysteme, die zusätzlich auf 3D-Strukturen, handverfasste evolutionäre Stammbäume oder Bevölkerungsdaten zurückgreifen. In klinischen Benchmarks, die aus der ClinVar-Datenbank für krankheitsassoziierte Varianten gezogen wurden, übertrifft das Hauptmodell von VESM viele weit verbreitete Werkzeuge und schlägt sogar AlphaMissense, ein jüngeres, viel beachtetes System, das Struktur- und Bevölkerungsdaten kombiniert. Entscheidend ist, dass VESM auch bei sehr seltenen Varianten gut performt — genau die Varianten, die Klinikerinnen und Kliniker am schwersten zu interpretieren finden. Die Modelle schneiden außerdem hervorragend bei Labormaßen ab, die testen, wie Mutationen Proteinfunktion, Stabilität und Bindung beeinflussen, und sie können die Größe und Richtung von Variantenwirkungen auf reale klinische Merkmale in großen Biobank-Datensätzen verfolgen.
Was das für Genetik und Medizin bedeutet
Durch das sorgfältige Kombinieren und Verfeinern der Stärken vieler Sequenzmodelle zeigt diese Arbeit, dass rohe Proteinsequenzen allein genug Signal tragen können, um die Auswirkungen genetischer Veränderungen auf dem Stand der Technik vorherzusagen. Das Ergebnis ist eine Familie von Werkzeugen, die einfacher einzusetzen sind als strukturlastige oder datenhungrige Systeme und gleichzeitig über Proteine von Menschen, Mikroben und Viren hinweg hohe Genauigkeit bewahren. Für Klinikerinnen und Kliniker sowie Forschende bedeutet das schnellere, verlässlichere Priorisierung von DNA-Varianten, eine klarere Einschätzung, wie stark eine Veränderung krankheitsrelevante Merkmale beeinflussen kann, und einen praktischen Weg, leistungsfähige Vorhersagemodelle in die tägliche Genetik- und Protein-Design-Praxis zu bringen.
Zitation: Dinh, T., Jang, SK., Zaitlen, N. et al. Compressing the collective knowledge of ESM into a single protein language model. Nat Methods 23, 772–784 (2026). https://doi.org/10.1038/s41592-026-03050-9
Schlüsselwörter: Protein-Sprachmodelle, Vorhersage von Variantenwirkungen, genetische Varianten, evolutionäre Signale, klinische Genomik