Clear Sky Science · en
Compressing the collective knowledge of ESM into a single protein language model
Why turning many protein models into one matters
Every person carries millions of tiny differences in their DNA, many of which change single building blocks in proteins. Most of these changes are harmless, but some can lead to disease. Doctors and researchers would love a fast, accurate way to tell which is which, using only the protein sequence. This article describes a new strategy that takes the collective know‑how of many existing protein “language models” and compresses it into a single, efficient system that rivals or beats today’s best tools for judging the impact of genetic changes.
From reading sentences to reading proteins
Protein language models borrow ideas from the technology that powers modern translation and chat systems. Instead of learning words in a sentence, they learn patterns in strings of amino acids, the letters of protein sequences. By training on hundreds of millions of natural proteins, these models pick up signals about which positions are highly conserved and which tolerate change. Those patterns can then be used to score genetic variants: if a change breaks a pattern that evolution strongly protects, it is more likely to be harmful. Until now, the strongest methods combined these models with extra information such as 3D structure or evolutionary family trees, making them powerful but also complex and sometimes hard to apply broadly.
Letting models teach each other
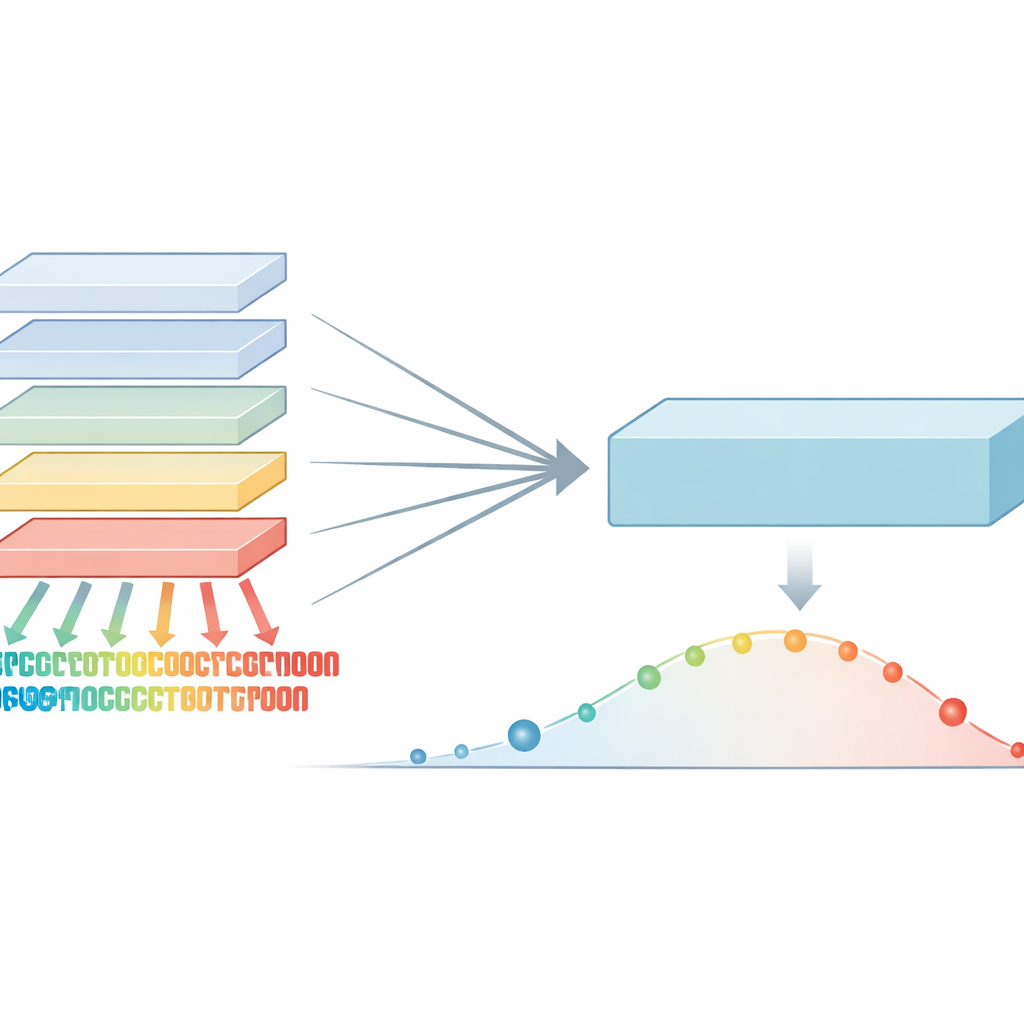
The authors noticed that different protein language models, even when built on similar data, have distinct strengths and blind spots. One model might be especially sharp at spotting sensitive regions in one protein family, while another excels elsewhere. Rather than averaging their opinions, the team introduced a “maximum-confidence” rule: for every possible protein change, they look across a whole family of models and keep whichever one is most certain the change is damaging. This step produces an enriched set of scores that captures the strongest evolutionary warning signals from any model in the group, instead of washing them out.
Boiling many voices down to one
Working with this enriched signal, the researchers designed a training process they call co-distillation. Here, all the original models repeatedly practice matching the strongest combined signal, taking turns as “student” and “teacher” depending on where each is most confident. In early rounds, the maximum‑confidence rule helps highlight subtle but important patterns that some models missed. In later rounds, a gentler averaging step helps the models agree and smooth out noise. Through several cycles, one large model gradually absorbs almost all of the useful information that was previously spread across the full ensemble. This final model, named VESM‑3B, is then used to train smaller versions that run fast enough for large genetic datasets while keeping most of the accuracy.
Beating complex systems with just sequences
Despite using only raw protein sequences as input, the VESM models match or surpass sophisticated competitors that also rely on 3D structures, hand‑built evolutionary histories, or data from human populations. On clinical benchmarks drawn from the ClinVar database of disease variants, the main VESM model outperforms many widely used tools and even edges out AlphaMissense, a recent high‑profile system that blends structure and population data. Crucially, VESM’s performance holds up for very rare variants, the ones clinicians most struggle to interpret. The models also excel on laboratory measurements that test how mutations affect protein fitness, stability and binding, and they can track the size and direction of variant effects on real clinical traits in large biobank datasets.
What this means for genetics and medicine
By carefully combining and refining the strengths of many sequence models, this work shows that raw protein sequences alone can carry enough signal to predict the impact of genetic changes at state‑of‑the‑art levels. The result is a family of tools that are simpler to deploy than structure‑heavy or data‑hungry systems, yet remain highly accurate across proteins from humans, microbes and viruses. For clinicians and researchers, this means faster, more reliable triage of DNA variants, a clearer sense of how strongly a change may influence disease‑related traits, and a practical way to bring powerful prediction models into everyday genetics and protein design workflows.
Citation: Dinh, T., Jang, SK., Zaitlen, N. et al. Compressing the collective knowledge of ESM into a single protein language model. Nat Methods 23, 772–784 (2026). https://doi.org/10.1038/s41592-026-03050-9
Keywords: protein language models, variant effect prediction, genetic variants, evolutionary signals, clinical genomics