Clear Sky Science · it
Comprimere la conoscenza collettiva di ESM in un singolo modello linguistico per proteine
Perché trasformare molti modelli proteici in uno solo è importante
Ogni persona porta con sé milioni di piccole differenze nel DNA, molte delle quali alterano singoli mattoni delle proteine. La maggior parte di questi cambiamenti è innocua, ma alcuni possono causare malattie. Medici e ricercatori desiderano uno strumento veloce e accurato per distinguere gli uni dagli altri usando soltanto la sequenza proteica. Questo articolo descrive una nuova strategia che raccoglie il sapere condiviso di molti modelli linguistici per proteine esistenti e lo comprime in un unico sistema efficiente che eguaglia o supera gli strumenti attuali più avanzati nell’interpretare l’impatto delle variazioni genetiche.
Dal leggere frasi al leggere proteine
I modelli linguistici per proteine prendono in prestito idee dalla tecnologia che alimenta i moderni sistemi di traduzione e chat. Invece di apprendere parole in una frase, apprendono schemi nelle stringhe di amminoacidi, le “lettere” delle sequenze proteiche. Allenandosi su centinaia di milioni di proteine naturali, questi modelli raccolgono segnali su quali posizioni sono fortemente conservate e quali tollerano cambiamenti. Questi schemi possono poi essere usati per valutare le varianti genetiche: se una modifica rompe un modello che l’evoluzione protegge con forza, è più probabile che sia dannosa. Finora, i metodi più potenti combinavano questi modelli con informazioni aggiuntive come la struttura 3D o gli alberi evolutivi di famiglia, rendendoli efficaci ma anche complessi e talvolta difficili da applicare in modo generalizzato.
Lasciare che i modelli si insegnino a vicenda
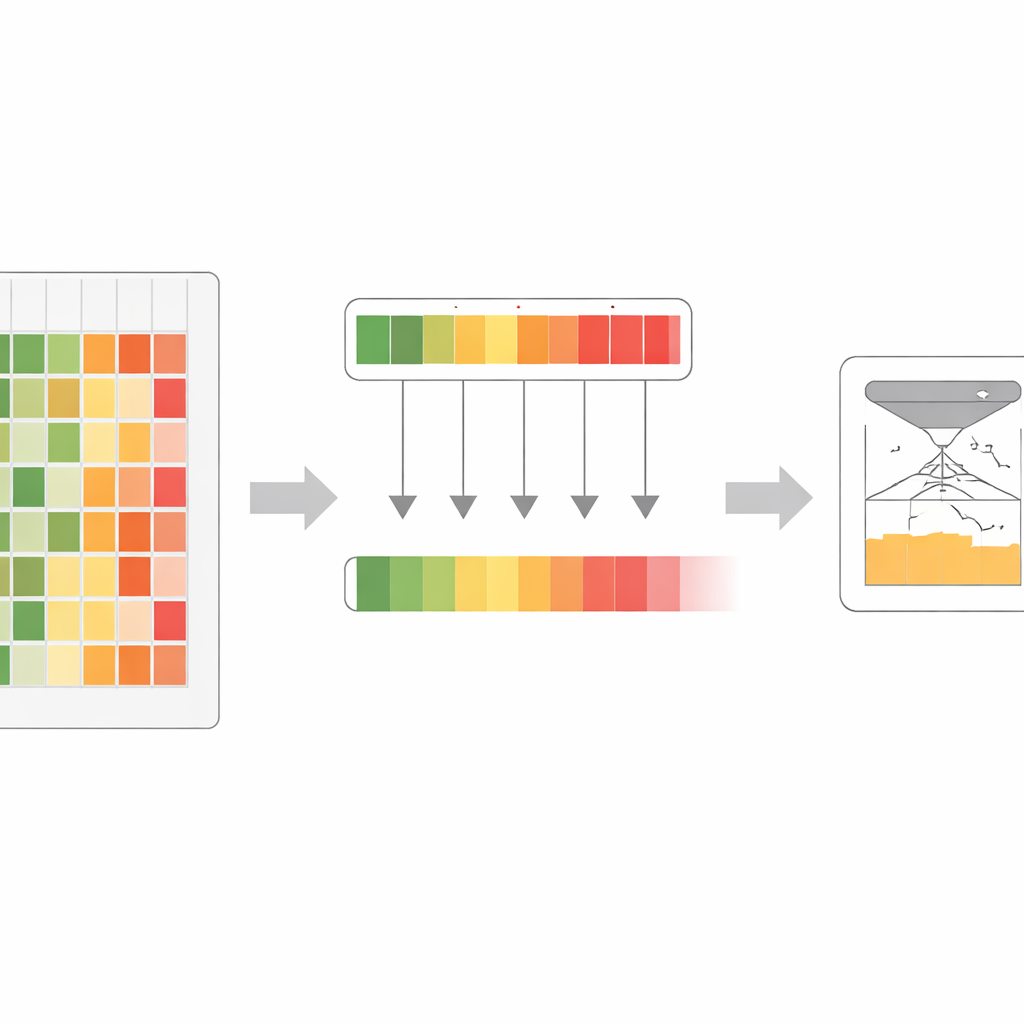
Gli autori hanno osservato che diversi modelli linguistici per proteine, anche se costruiti su dati simili, mostrano punti di forza e limiti differenti. Un modello può essere particolarmente abile nell’individuare regioni sensibili in una famiglia proteica, mentre un altro eccelle altrove. Invece di mediare le loro opinioni, il gruppo ha introdotto una regola di “massima confidenza”: per ogni possibile cambiamento proteico, esaminano l’intera famiglia di modelli e conservano quello che è più certo che la variante sia dannosa. Questo passaggio produce un insieme di punteggi arricchito che cattura gli avvisi evolutivi più forti provenienti da qualunque modello del gruppo, invece di diluirli.
Ridurre molte voci in una sola
Lavorando con questo segnale arricchito, i ricercatori hanno progettato un processo di addestramento che chiamano co-distillazione. Qui, tutti i modelli originali si esercitano ripetutamente a riprodurre il segnale combinato più forte, alternandosi nei ruoli di “studente” e “insegnante” a seconda di dove ciascuno è più sicuro. Nelle prime fasi, la regola della massima confidenza aiuta a evidenziare schemi sottili ma importanti che alcuni modelli avevano perso. Nelle fasi successive, un passaggio di mediazione più morbido favorisce l’accordo tra i modelli e smussa il rumore. Attraverso diversi cicli, un grande modello assorbe gradualmente quasi tutte le informazioni utili che prima erano disperse nell’intero insieme. Questo modello finale, denominato VESM‑3B, viene poi usato per addestrare versioni più piccole che girano abbastanza veloci per grandi insiemi di dati genetici mantenendo gran parte dell’accuratezza.
Battere sistemi complessi usando solo le sequenze
Nonostante usino soltanto le sequenze proteiche grezze come input, i modelli VESM eguagliano o superano concorrenti sofisticati che si basano anche su strutture 3D, storie evolutive costruite a mano o dati sulle popolazioni umane. Su benchmark clinici estratti dal database ClinVar delle varianti patologiche, il modello principale VESM supera molti strumenti largamente usati e persino supera AlphaMissense, un recente sistema di alto profilo che combina struttura e dati di popolazione. È fondamentale che le prestazioni di VESM rimangano solide anche per varianti molto rare, quelle che i clinici faticano maggiormente a interpretare. I modelli eccellono anche nelle misurazioni di laboratorio che testano come le mutazioni influenzano fitness, stabilità e legame delle proteine, e riescono a seguire l’entità e la direzione degli effetti delle varianti su tratti clinici reali in grandi set di biobanche.
Cosa significa per la genetica e la medicina
Combinando e perfezionando con cura i punti di forza di molti modelli basati sulle sequenze, questo lavoro dimostra che le sole sequenze proteiche grezze possono contenere segnali sufficienti a prevedere l’impatto delle alterazioni genetiche a livelli all’avanguardia. Il risultato è una famiglia di strumenti più semplice da distribuire rispetto a sistemi che richiedono strutture o grandi quantità di dati, ma che rimane altamente accurata su proteine umane, microbiche e virali. Per clinici e ricercatori, ciò significa una selezione più rapida e affidabile delle varianti del DNA, una comprensione più chiara di quanto fortemente un cambiamento possa influenzare tratti legati a malattie e un modo pratico per integrare modelli predittivi potenti nei flussi di lavoro quotidiani di genetica e progettazione proteica.
Citazione: Dinh, T., Jang, SK., Zaitlen, N. et al. Compressing the collective knowledge of ESM into a single protein language model. Nat Methods 23, 772–784 (2026). https://doi.org/10.1038/s41592-026-03050-9
Parole chiave: modelli linguistici per proteine, predizione dell'effetto delle varianti, varianti genetiche, segnali evolutivi, genomica clinica