Clear Sky Science · fr
Compresser le savoir collectif des ESM en un seul modèle de langage protéique
Pourquoi transformer de nombreux modèles protéiques en un seul est important
Chaque personne porte des millions de petites différences dans son ADN, dont beaucoup modifient des acides aminés individuels dans les protéines. La plupart de ces changements sont inoffensifs, mais certains peuvent entraîner des maladies. Les médecins et les chercheurs souhaiteraient disposer d’un moyen rapide et précis pour distinguer l’un de l’autre en se basant uniquement sur la séquence protéique. Cet article décrit une nouvelle stratégie qui rassemble le savoir collectif de nombreux modèles « de langage » protéiques existants et le compresse dans un système unique et efficace, capable d’égaler ou de surpasser les meilleurs outils actuels pour évaluer l’impact des changements génétiques.
De la lecture de phrases à la lecture de protéines
Les modèles de langage protéique empruntent des idées à la technologie qui alimente les systèmes modernes de traduction et de conversation. Plutôt que d’apprendre des mots dans une phrase, ils apprennent des motifs dans des chaînes d’acides aminés, les « lettres » des séquences protéiques. En s’entraînant sur des centaines de millions de protéines naturelles, ces modèles détectent des signaux indiquant quelles positions sont fortement conservées et lesquelles tolèrent les variations. Ces motifs peuvent ensuite servir à scorer des variantes génétiques : si une modification rompt un motif fortement protégé par l’évolution, elle a plus de chances d’être délétère. Jusqu’ici, les méthodes les plus performantes combinaient ces modèles avec des informations supplémentaires, comme la structure 3D ou des arbres évolutifs, ce qui les rendait puissantes mais aussi complexes et parfois difficiles à appliquer de façon générale.
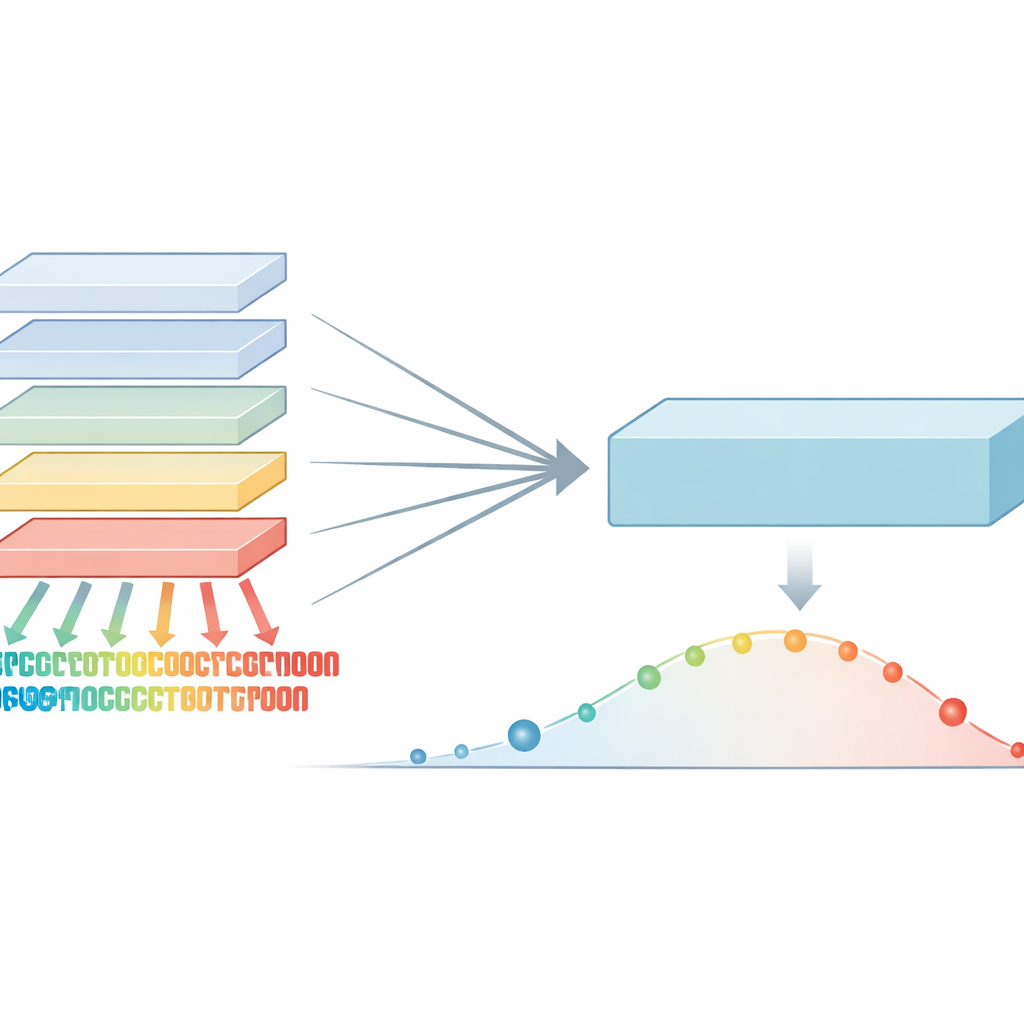
Laisser les modèles s’enseigner entre eux
Les auteurs ont observé que différents modèles de langage protéique, même s’ils sont entraînés sur des données similaires, présentent des forces et des angles morts distincts. Un modèle peut être particulièrement performant pour repérer des régions sensibles dans une famille de protéines, tandis qu’un autre excelle ailleurs. Plutôt que de moyenniser leurs avis, l’équipe a introduit une règle de « confiance maximale » : pour chaque changement protéique possible, ils examinent l’ensemble des modèles et conservent celui qui est le plus certain que la modification est dommageable. Cette étape produit un ensemble de scores enrichis qui capture les signaux d’alerte évolutifs les plus forts provenant de n’importe quel modèle du groupe, au lieu de les diluer.
Réduire plusieurs voix à une seule
En travaillant à partir de ce signal enrichi, les chercheurs ont conçu un processus d’entraînement qu’ils appellent co-distillation. Ici, tous les modèles originaux s’exercent à reproduire à plusieurs reprises le signal combiné le plus fort, en prenant tour à tour les rôles d’« élève » et de « professeur » selon l’endroit où chacun est le plus confiant. Lors des premières itérations, la règle de confiance maximale permet de mettre en évidence des motifs subtils mais importants que certains modèles avaient manqués. Dans les phases ultérieures, une étape d’agrégation plus douce aide les modèles à se mettre d’accord et à lisser le bruit. Au fil de plusieurs cycles, un grand modèle absorbe progressivement presque toute l’information utile qui était auparavant répartie dans l’ensemble. Ce modèle final, nommé VESM‑3B, sert ensuite à entraîner des versions plus petites qui s’exécutent assez rapidement pour traiter de larges jeux de données génétiques tout en conservant la majeure partie de la précision.
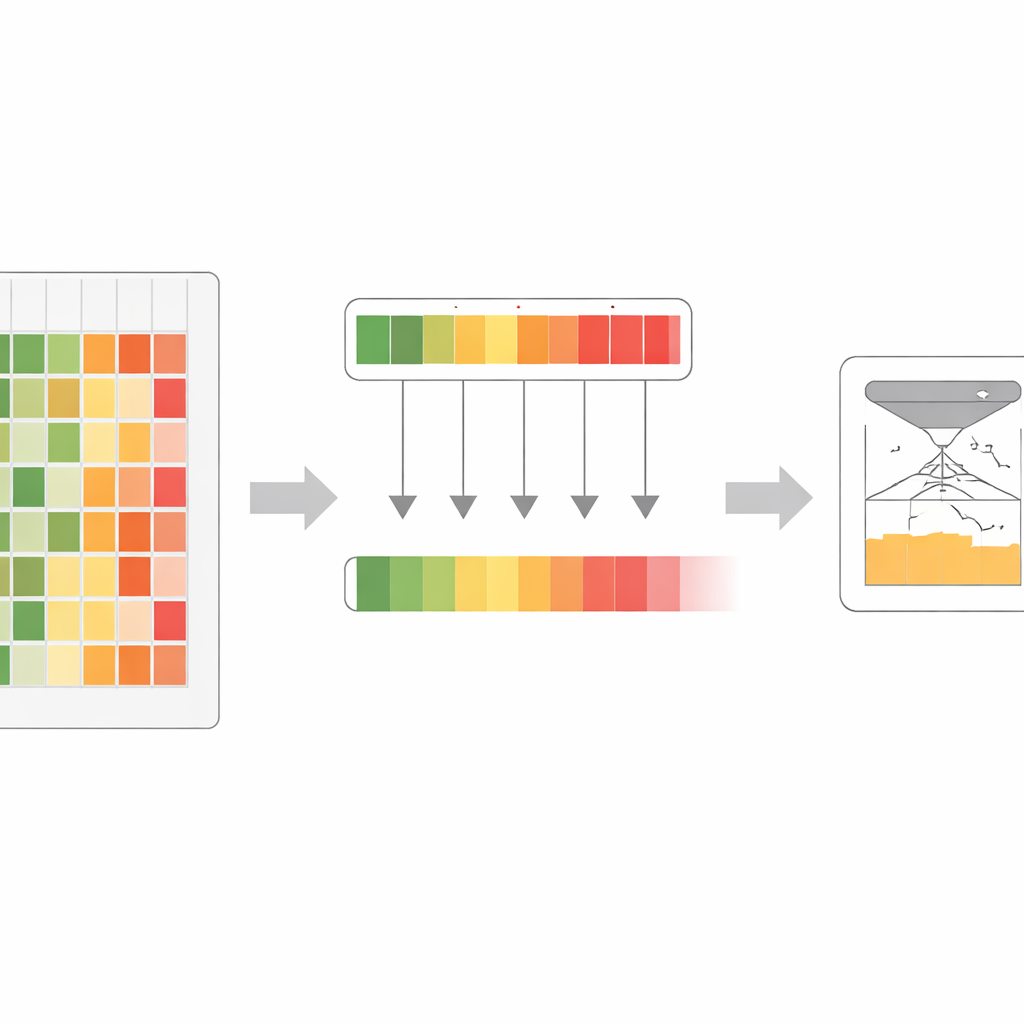
Battre des systèmes complexes en n’utilisant que les séquences
Malgré l’utilisation uniquement de séquences protéiques brutes en entrée, les modèles VESM égalent ou surpassent des concurrents sophistiqués qui s’appuient également sur des structures 3D, des histoires évolutives manuellement construites ou des données de populations humaines. Sur des références cliniques issues de la base ClinVar des variantes pathogènes, le modèle principal VESM surpasse de nombreux outils largement utilisés et devance même AlphaMissense, un système récent et très médiatisé qui combine structure et données de population. Surtout, les performances de VESM se maintiennent pour les variantes très rares, celles que les cliniciens peinent le plus à interpréter. Les modèles excellent aussi sur des mesures expérimentales en laboratoire qui évaluent l’impact des mutations sur la fitness, la stabilité et l’affinité des protéines, et ils peuvent suivre la taille et la direction des effets des variantes sur des traits cliniques réels dans de larges bases de données de biobanques.
Ce que cela signifie pour la génétique et la médecine
En combinant et en affinant soigneusement les forces de nombreux modèles de séquence, ce travail montre que les seules séquences protéiques brutes peuvent porter suffisamment d’information pour prédire l’impact des changements génétiques à des niveaux de pointe. Le résultat est une famille d’outils plus simples à déployer que les systèmes exigeant la structure ou de vastes quantités de données, tout en restant très précis sur des protéines humaines, microbiennes et virales. Pour les cliniciens et les chercheurs, cela signifie un triement des variantes d’ADN plus rapide et plus fiable, une estimation plus claire de la force avec laquelle une modification peut influencer des traits liés à la maladie, et un moyen pratique d’intégrer des modèles prédictifs puissants dans les flux de travail quotidiens de la génétique et du design protéique.
Citation: Dinh, T., Jang, SK., Zaitlen, N. et al. Compressing the collective knowledge of ESM into a single protein language model. Nat Methods 23, 772–784 (2026). https://doi.org/10.1038/s41592-026-03050-9
Mots-clés: modèles de langage protéique, prévision des effets de variantes, variantes génétiques, signaux évolutifs, génomique clinique